Études de la propagation de la protéine huntingtine mutée - Mémoire Philippe Gosset Maîtrise en médecine moléculaire - avec mémoire - Corpus UL
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Études de la propagation de la protéine huntingtine
mutée
Mémoire
Philippe Gosset
Maîtrise en médecine moléculaire - avec mémoire
Maître ès sciences (M. Sc.)
Québec, Canada
© Philippe Gosset, 2020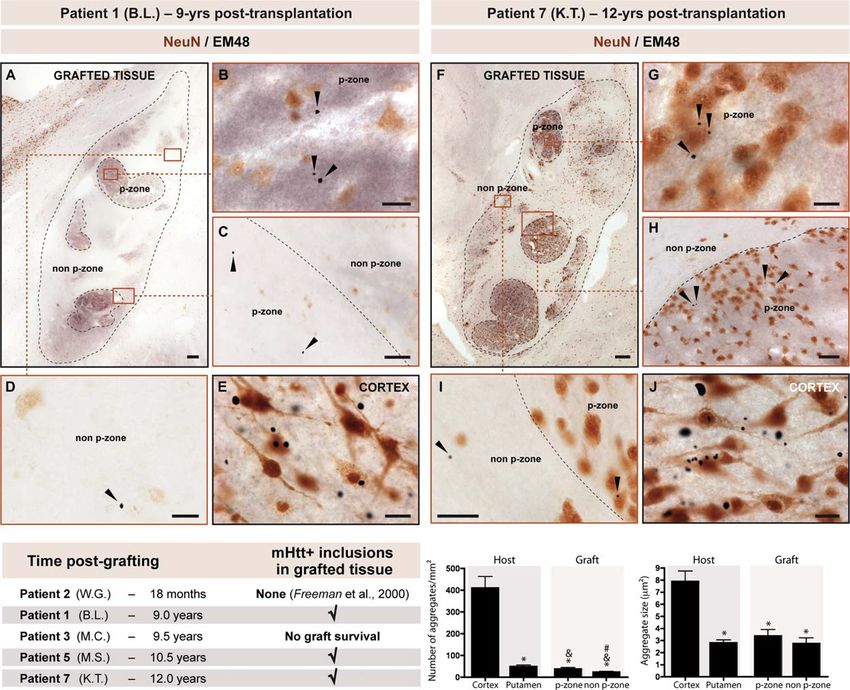
Etudes de la propagation de la protéine huntingtine
mutée
Mémoire
Philippe Gosset
Sous la direction de :
Francesca Cicchetti, directeur ou directrice de recherche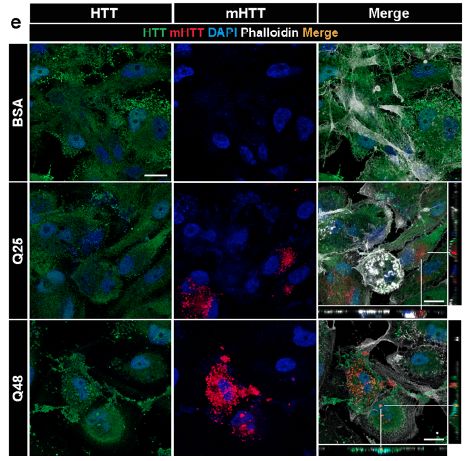
Résumé
La maladie de Huntington (MH) est une maladie neurodégénérative d’origine
génétique entrainant la mutation de la protéine huntingtine (HTT). Cette protéine
essentielle, sous sa forme mutée (mHTT) s’agrège, lui conférant ainsi sa toxicité. Au niveau
cérébral, la MH se caractérise par une mort neuronale accrue entrainant des troubles
moteurs, cognitifs et psychiatriques.
Notre projet de recherche porte sur la caractérisation de certaines particularités de la
MH, en particulier sur les facultés de cette pathologie à se propager entre espèces. Cette
particularité s’inscrit dans une hypothèse proposant que la MH soit incluse dans la
catégorie des maladies à prions, bien que cette théorie ne fasse pas encore l’unanimité au
sein de la communauté scientifique. A travers celui-ci, nous avons cherché à déterminer si
la mHTT présente dans les homogénats cérébraux des patients MH, peut induire une
pathologie chez différents modèles animaux. Pour cela, nous avons effectué trois protocoles
distincts où des homogénats ont été injectés dans le cerveau de souris adultes a) de type
sauvage et b) de type BACHD ou c) de primates non-humains.
Les résultats obtenus de cette étude semblent indiquer que la mHTT dérivée du
cerveau humain a une capacité limitée à se propager entre les cellules et ne représente pas
des caractéristiques de type prion. Ces observations contrastent avec de précédents travaux
démontrant que d'autres formes de mHTT (fibrilles, fibroblastes ou cellules souches
pluripotentes induites dérivées de cas de MH) peuvent en effet disséminer la maladie dans
le cerveau à la manière d'un prion.
ii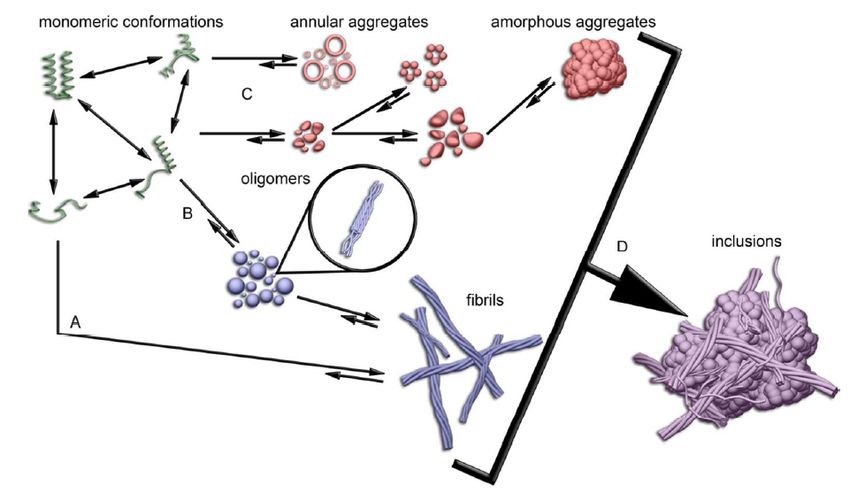
Abstract
Huntington's disease (HD) is a neurodegenerative disease caused by a mutation of
the gene coding for the huntingtin protein (HTT). In its mutated form (mHTT), this
essential protein forms into cellular aggregates, conferring toxicity to the new entity. HD is
also characterized by marked neuronal death which underlies the motor, cognitive and
psychiatric symptoms known to the pathology.
The research project presented herein focused on investigating the ability of the
mHTT to spread between cellular elements. This is part of a larger research program which
proposes that HD may be, as other neurodegenerative diseases, a prion-like disorder. We
therefore sought to determine whether the mHTT found in homogenates derived from post-
mortem brain tissue of HD patients can induce pathology after injection in different animal
models. For this, we carried out three separate protocols: homogenates were injected into
the brains of adult a) wild type and b) BACHD mice or c) non-human primates.
The results of this study indicate that mHTT derived from post-mortem brain tissue
of HD patients has a limited ability to spread between cells and does not represent prion-
like characteristics. These observations contrast with previous work demonstrating that
other forms of mHTT (fibrils, fibroblasts or induced pluripotent stem cells derived from
HD cases) can indeed disseminate disease in a prion-like fashion as shown in various in
vitro and in vivo models.
iii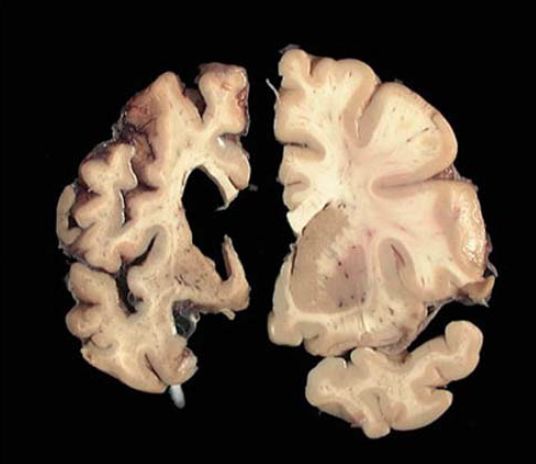
Table des matières
Résumé ............................................................................................................................................................... ii
Abstract ..............................................................................................................................................................iii
Table des matières ........................................................................................................................................... iv
Liste des figures ............................................................................................................................................... vi
Liste des abréviations, sigles, acronymes ....................................................................................................vii
Remerciements ................................................................................................................................................. ix
Avant-propos ..................................................................................................................................................... x
Introduction ....................................................................................................................................................... 1
La maladie de Huntington ........................................................................................................................... 1
Historique .................................................................................................................................................. 1
Epidémiologie .......................................................................................................................................... 1
Symptômes................................................................................................................................................ 2
Etiologie .................................................................................................................................................... 3
Atteinte physiologique ............................................................................................................................ 4
Traitements disponibles et envisagés .................................................................................................... 5
Modèles animaux ..................................................................................................................................... 6
La protéine Huntingtine............................................................................................................................... 7
Caractéristiques ........................................................................................................................................ 7
Généralités ................................................................................................................................................ 7
Rôle de la HTT ......................................................................................................................................... 9
La HTT mutée ........................................................................................................................................ 11
La protéine prion ........................................................................................................................................ 13
Propagation de la mHTT ........................................................................................................................... 14
In vitro ..................................................................................................................................................... 14
In vivo ...................................................................................................................................................... 17
Modèles animaux à homogénats .......................................................................................................... 19
Projet de recherche ..................................................................................................................................... 20
Chapitre 1 Evidence for the spread of human-derived mutant huntingtin protein in mice and non-
human primates ............................................................................................................................................... 21
1.1 Résumé .................................................................................................................................................. 23
1.2 Abstract ................................................................................................................................................. 24
1.3 Introduction .......................................................................................................................................... 25
iv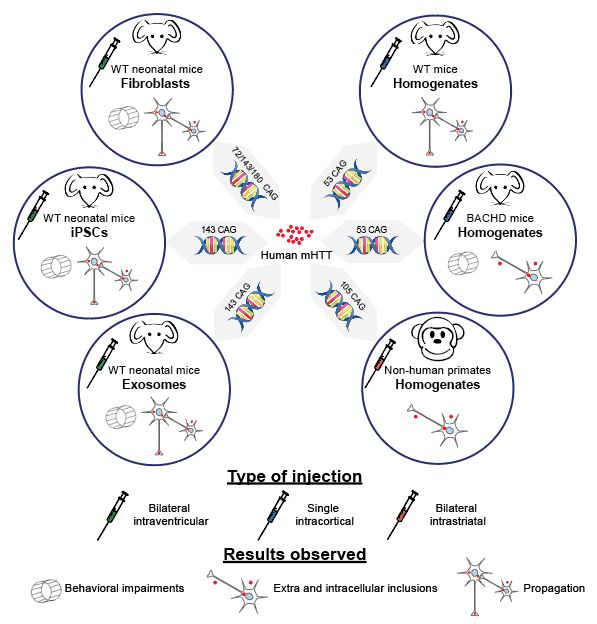
1.4 Material and Methods ......................................................................................................................... 27
1.5 Results ................................................................................................................................................... 35
1.6 Discussion ............................................................................................................................................. 40
1.7 Competing interests ............................................................................................................................. 44
1.8 Acknowledgments ............................................................................................................................... 44
1.9 Author contributions ........................................................................................................................... 44
1.10 Figure legends .................................................................................................................................... 45
1.11 References .......................................................................................................................................... 50
Conclusion ....................................................................................................................................................... 53
Démonstrations des capacités de propagation de la mHTT ................................................................. 53
Améliorations possibles de l’étude .......................................................................................................... 58
Perspectives ................................................................................................................................................. 59
Perspectives futures d’étude de la mHTT extracellulaire dans le système nerveux central et la
circulation générale .................................................................................................................................... 59
Bibliographie ................................................................................................................................................... 62
v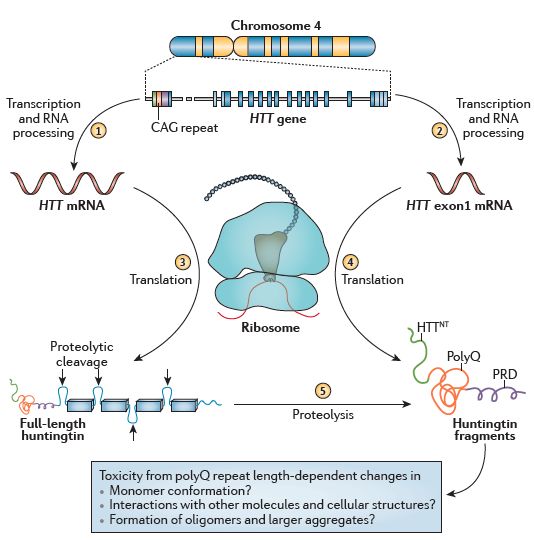
Liste des figures
Figure 0.1 : Coupes coronales de télencéphales humains.
Figure 0.2 : Structure du gène de la huntingtine humaine.
Figure 0.3 : Structure et transformation de la HTT.
Figure 0.4: Modèle schématique illustrant le mauvais repliement et l'agrégation de
protéines contenant une queue polyQ étendue.
Figure 0.5: Marquages illustrant les propriétés d’interaction de la mHTT avec la HTT
endogène.
Figure 0.6: Immunomarquages démontrant la présence de mHTT dans les greffons
chez un patient huntingtonien – 12 ans après transplantation.
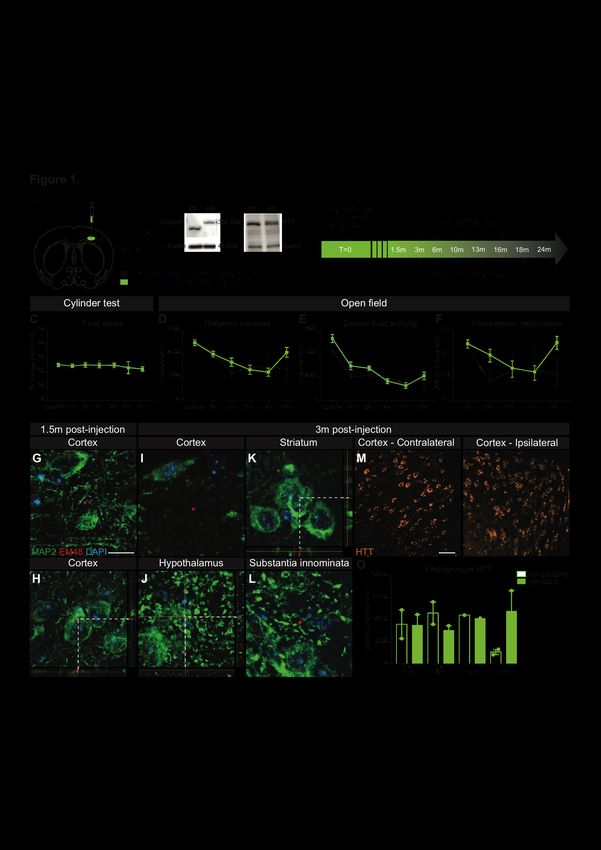
Figure 1.1. Cortical injection of human mHTT homogenates in WT mice.
Figure 1.2. Cortical injection of human mHTT homogenates in BACHD mice.
Figure 1.3. Striatal injection of human mHTT homogenates in non-human primates.
Figure 0.7 : Schéma récapitulatif et comparatif de différentes études portant sur la
propagation de la mHTT.
vi
Liste des abréviations, sigles, acronymes
APP amyloid precursor protein / protéine précurseur de l’amyloide
ARN acide ribonucléique
ARNm acide ribonucléique messager
BDNF Brain-Derived Neurotrophic Factor / facteur neurotrophique dérivé du cerveau
CAG Cytosine Adénosine Guanine
CSPi cellules souches pluripotentes induites
DRP domaine enrichi en proline
GPe globus pallidus externe
GPi globus pallidus interne
HTT protéine huntingtine
HTT gène de la protéine huntingtine
LCR Liquide céphalo-rachidien
LRP1 protéine associée aux récepteurs des lipoprotéines 1
MA Maladie d’Alzheimer
MH Maladie de Huntington
mHTT protéine huntingtine mutée
MP Maladie de Parkinson
MSN neurones épineux moyens
NCOR co-répresseur nucléaire
NES signal d’export nucléaire
viiNF-kB nuclear factor-kB
P53 protéine 53
polyQ polyglutamine
PrPc protéine prion cytoplasmique
PrPsc PrP scrapie
PSN polymorphismes d’un seul nucléotide
SN substantia nigra
SNC système nerveux central
STN noyau subthalamique
WT Wild-Type / sauvage
viiiRemerciements
Je tiens à remercier la Dre Francesca Cicchetti de m’avoir accepté dans son
laboratoire. Elle m’a permis de mener des travaux de recherche au sein de son laboratoire et
je la remercie plus particulièrement pour ses conseils, son écoute et son temps consacré à la
réalisation de ce travail expérimental et de l’article figurant dans ce mémoire.
Je remercie également l’ensemble de l’équipe Cicchetti : à savoir les étudiants et
notre professionnelle de recherche pour leur accueil au sein du laboratoire, leur bonne
humeur ainsi que leurs conseils.
ixAvant-propos
Au cours de ma maitrise dans le laboratoire de la Dre Francesca Cicchetti, j’ai eu la
chance de participer à plusieurs projets de recherche sur la propagation de la protéine
huntingtine mutée (mHTT) dans la maladie de Huntington. Cependant, j’ai principalement
axé mon temps sur un de ceux-ci : projet qui est présenté dans le chapitre 1 puisqu’il fait
l’objet d’une publication où je suis premier auteur. Par ailleurs, j’ai traité les données
comportementales et préparé le plan expérimental d’analyse post-mortem d’une étude que
j’avais commencé à mener: cette étude porte sur l’injection de fibroblastes humains
contenant des protéines pathologiques (mHTT) dans le cerveau de singes adultes et bébés.
Aussi, j’ai contribué à un projet sur la propagation de la mHTT entre deux souris couplées
en parabiose dont l’une est traitée par irradiation. Enfin, j’ai effectué les analyses de
génotypes chez des souris résultants de croisements génétiques pour observer la
propagation de la mHTT chez un nouveau modèle de souris transgéniques zQ175/LRP1.
L’article « Evidence for the spread of human-derived mutant huntingtin protein in
mice and non-human primates » présenté au chapitre 1 a été accepté pour publication en
2020 dans le journal Neurobiology of Disease. Cet article a été réalisé en collaboration avec
Erwan Bézard en France (Université de Bordeaux, Institut des maladies
neurodégénératives, UMR 5293, Bordeaux, France, CNRS UMR 5293) et son équipe en
Chine (Institute of Laboratory Animal Sciences, China Academy of Medical Sciences,
Beijing, China).
Je suis premier auteur de cet article pour avoir réalisé les analyses des données
comportementales de tous les animaux, les analyses post-mortem de chaque groupe et pour
avoir réalisé les différentes figures et la première version du manuscrit.
La liste complète des auteurs est : *1Philippe Gosset, *1Alexander Maxan, *1Melanie
Alpaugh, 2Ludivine Breger, 2Benjamin Dehay, 4Giulia Cisbani, 1Nadia Fortin, 3Zhu Tao,
3
Zhang Ling, 3Chuan Qin, 4Jean Paul G. Vonsattel, 1,5
Steve Lacroix, 1,5
Abid Oueslati,
2 1,6
Erwan Bezard and Francesca Cicchetti.
xAffiliations :
1
Centre de Recherche du CHU de Québec - Université Laval, Axe Neurosciences, Québec,
QC, Canada, G1V 4G2; 2Université de Bordeaux, Institut des maladies neurodégénératives,
UMR 5293, Bordeaux, France, CNRS UMR 5293; 3Institute of Laboratory Animal
Sciences, China Academy of Medical Sciences, Beijing, China; 4University of Toronto,
Department of Nutritional Sciences, Toronto, ON, Canada, M5S 1A8; 5New-York Brain
Bank, Columbia University, New York, NY, United States, 10032; 6Département de
Médicine Moléculaire, Université Laval, Québec, QC, Canada, G1K 0A6; 7Département de
Psychiatrie & Neurosciences, Université Laval, Québec, QC, Canada, G1K 0A6
*Égale contribution.
xiIntroduction
La maladie de Huntington
Historique
En 1872, le médecin américain Georges Huntington publie pour la première fois
dans le journal scientifique Medical and Surgical Reporter, la description de la maladie de
Huntington (MH) (Huntington 1872). Dans cet article, on peut noter que la caractéristique
majeure de la pathologie se présente sous forme de mouvements involontaires ; ceux-ci
évoquant une sorte de danse effectuée par les malades : « la chorée ». Le terme de
« chorée », provenant du latin choreus qui signifie une danse dans l’Antiquité, a alors été
choisi pour définir la maladie. A travers son observation, le docteur Huntington suppose la
nature héréditaire et dominante de la MH: en effet, lorsqu’un parent présente la pathologie,
un ou plusieurs enfants, avec une probabilité de 50%, la développe à son tour ; et lorsqu’un
de ces enfants qui n’a pas hérité de la maladie a à son tour des enfants, ceux-ci ne
présentent pas la MH. En 1883, Westphal décrit des symptômes juvéniles ressemblant à
ceux observés dans la MH mais les a attribués à une cause autre que la MH. En effet, les
patients présentaient une prédominance d'hypokinésie et de rigidité ainsi, le terme
« variante de Westphal » était souvent utilisé pour décrire le tableau clinique de la MH
juvénile. Bien qu'il y ait eu plusieurs rapports sur la neuropathologie de la MH, ce n'est que
dans les années 1920 qu'il y a eu un accord que dans la MH, les changements dans le
cerveau sont principalement dégénératifs et atrophiques, et que la structure du noyau caudé
est la plus affectée (G. Bates, Harper, and Jones 2002). Enfin, c’est seulement en 1993 que
le Huntington’s Disease Collaborative Research Group identifie le gène responsable de la
maladie (HDCRG, 1993). Ce gène est d’abord appelé IT15 (Interesting Gene 15) puis
renommé « huntingtine » (MacDonald et al. 1993).
Epidémiologie
La prévalence de la MH semble être très variable sur le globe ; c’est pourquoi des
revues de la littérature et des méta-analyses ont été établies. Ainsi, dans une méta-analyse
rassemblant 8 articles originaux étudiant l’incidence et 17 examinant la prévalence, il nous
est rapporté que l’incidence est de 0.38 nouveaux cas pour 100 000 personnes chaque année
et que la prévalence mondiale de la MH serait de 2.71 cas pour 100 000 individus.
1Cependant, on peut noter une forte hétérogénéité dans les chiffres publiés concernant la
prévalence à travers les différentes études entre les populations asiatiques, européennes et
nord-américaines par exemple. En effet, la prévalence en Europe, Amérique du Nord et
Australie est de 5.7 cas pour 100 000 individus mais seulement de 0.4 en Asie (Pringsheim
et al. 2012). Néanmoins, si on ne s’intéresse qu’à la prévalence de la MH au Canada, on
remarque qu’elle est beaucoup plus élevée que la moyenne avec 13.7 cas pour 100 000
canadiens (Fisher and Hayden 2014). Les différences observées pourraient être dues à des
différences génétiques au sein des différentes populations mondiales, portant sur le gène
impliqué dans la MH (voir plus bas dans le paragraphe « Etiologie »).
Symptômes
La MH, dans la plupart des cas, se déclare autour de 50ans ; cependant, on peut
noter quelques cas de formes juvéniles ou tardives (Flier et al. 1986; Phillips, Shannon, and
Barker 2008; Papp, Kaplan, and Snyder 2011). Elle comporte plusieurs phases : une phase
présymptomatique, qui précède l’apparition des symptômes ; une phase prodromique, qui
dure environ 15ans au cours desquels les patients présentent de légères altérations des
fonctions cognitives et motrices ; puis la phase déclarative de la maladie, qui permet le
diagnostic définitif puisqu’elle est marquée par l’apparition des symptômes moteurs
spécifiques et d’un test génétique et/ou de l’évaluation des antécédents familiaux du patient
(Reilmann, Leavitt, and Ross 2014). Dans cette dernière phase, on observe une
accentuation des symptômes cognitifs, moteurs et psychiatriques entrainant le décès du
patient. Les malades souffrent alors de mouvements involontaires (chorée), de bradykinésie
(ralentissement et perte de finesse dans le mouvement) et de l’altération de la coordination.
On observe des troubles de l’équilibre et de la posture ajoutés à des défauts d’articulation et
de déglutition. Sur le plan cognitif, on retrouve des troubles de l’attention, des pertes de
mémoire, un défaut d’orientation puis à un stade plus avancé, la démence privant alors le
patient de son autonomie (Peavy et al. 2010). Des troubles psychiatriques sont également
observables chez les patients atteints de le MH : les plus courant étant la dépression,
l’irritabilité, l’apathie et l’impulsivité (van Duijn et al. 2014; J. C. Thompson et al. 2012).
2Etiologie
La MH est considérée comme étant une maladie neurodégénérative mais c’est la
seule qui est d’origine purement génétique. Elle est causée par une mutation du gène
localisée dans la région 4p16.3 de l’exon 1 du chromosome 4 codant pour la protéine
huntingtine (HTT). En effet, la HTT possède une queue polyglutamine (polyQ) ; répétition
du triplet nucléotidique CAG (C : cytosine ; A : adénosine ; G : guanine) en N-terminal, et
sa longueur détermine la conformation de la HTT : donc de sa pathogénicité (Duyao et al.
1993; Gusella et al. 1983; Labbadia and Morimoto 2013). Ainsi, une queue polyQ
comprenant entre 6 et 36 triplets CAG permettra une fonction normale de la HTT. Au-delà
de 36 répétitions CAG, la protéine change de conformation et s’agrège : c’est la protéine
huntingtine mutée (mHTT). La pénétrance de la maladie est limitée dans le cas de 36 à 40
répétitions CAG, mais est complète une fois passé ce nombre (Kay et al. 2016). Nota bene,
un nombre de répétitions CAG supérieur à 60 est appelé « forme juvénile » du fait de
l’apparition précoce de la pathologie et de sa sévérité. L’allèle muté dans la MH est dit
« dominant », c’est-à-dire que sa présence en un seul exemplaire est suffisante pour
déclarer la pathologie (Conneally 1984). Ainsi, un enfant issu d’un couple où l’un des
parents est porteur de l’allèle muté présente une probabilité de 50% d’être porteur de la
mutation et donc de déclarer la MH à son tour. Malgré l’origine génétique, environ 10%
des cas de la MH sont sporadiques : c’est-à-dire que la cause n’est pas due à une mutation
héritée mais due à une mutation de novo (Falush et al. 2001; Sunwoo, Lee, and Kim 2010).
Cette origine génétique explique en partie l’épidémiologie variable de la MH à
travers le monde. Plusieurs théories sont avancées pour expliquer les différences observées.
Premièrement, la probabilité de développer la mutation de novo est directement corrélée à
la taille de la répétition CAG dans le gène huntingtine (HTT). Ainsi, il a été montré que
celle-ci n’était pas la même entre les populations européennes (18.4 triplets CAG en
moyenne) comparée aux populations asiatiques et africaines (16.4 triplets CAG) (Squitieri
et al. 1994). Deuxièmement, il existe des polymorphismes d’un seul nucléotide (PSN)
caractéristiques de différentes versions du gène HTT concordant avec la présence
d’expansion de CAG ; ceux-ci sont retrouvés, le plus souvent, dans les populations
européennes (Warby et al. 2009). Enfin, la troisième explication porterait sur une région
riche en CCG adjacente aux répétitions CAG. En effet, cette région est variable selon
3l’expression de certains allèles associés à l’apparition de la mutation de novo ; allèles
retrouvés dans les populations d’origine européenne (Pêcheux et al. 1995). Au contraire,
dans les populations asiatiques, on observe l’existence d’allèles ayant un effet protecteur
envers l’apparition de la mutation (Warby et al. 2011; Baine et al. 2013).
Atteinte physiologique
Sur le plan anatomique, la MH se caractérise par l’atteinte du striatum, une atrophie
grandissant tout au long de la maladie. Au sein des différentes populations de neurones que
l’on peut retrouver dans le striatum, ce sont les neurones de projections GABAergiques ;
les neurones épineux moyens (MSN), qui sont les plus touchés entrainant alors une
altération irréversible des réseaux neuronaux (Han et al. 2010; Tepper, Koós, and Wilson
2004). Le striatum est une structure cérébrale sous-corticale appartenant aux ganglions de
la base. Il comprend le noyau caudé, le putamen, le pallidum et le noyau accumbens, et est
impliqué dans de nombreuses fonctions dont l’initiation ou l’inhibition du mouvement
volontaire et involontaire ; ceci expliquant les symptômes moteurs caractéristiques de la
MH (Kravitz and Kreitzer 2012). D’autres zones sont également touchées par la
neurodégénérescence telles que le cortex, le cervelet, l’hippocampe, la substance noire ou
encore les noyaux du tronc cérébral (Paulson and Albin 2011). Bien que la MH soit
considérée comme une maladie neurodégénérative, elle ne touche pas seulement le système
nerveux central (SNC). En effet, le gène muté codant pour la protéine huntingtine (mHTT)
étant exprimé de façon ubiquitaire dans les cellules de l’organisme, cette mutation
occasionne des atteintes de différents organes périphériques.
Sur le plan cellulaire, l’expression de la mHTT entraine la formation d’agrégats
protéiques au sein du corps cellulaire, du noyau mais également dans le milieu
extracellulaire (Gutekunst et al. 1999). L’impact de ces agrégats sur les neurones et sur la
mort cellulaire n’est pas encore clairement établi. D’après la littérature, les agrégats
pourraient adopter différentes conformations dont certaines seraient toxiques ; ainsi, ils ne
seraient pas systématiquement nocifs pour la cellule hôte. D’ailleurs, on ne retrouve que
très peu d’agrégats dans les neurones striataux malgré le fait qu’il s’agisse de la région la
plus impactée de la MH (Hoffner and Djian 2015).
4Figure 0.1 : Coupes coronales de télencéphales humains. A droite, on observe le cerveau
d’un sujet sain et à gauche, celui d’un patient avec une atteinte avancée de la MH. Une
atrophie du cortex ainsi que du noyau caudé résultant en un élargissement du ventricule
sont notables sur le télencéphale du patient MH. D’après (Anton Reiner, Dragatsis, and
Dietrich 2011).
Traitements disponibles et envisagés
Il existe actuellement plusieurs traitements médicamenteux disponibles pour les
patients atteints de la MH cependant, il est important de noter que de ceux-ci, aucun n’est
curatif. De plus, seul un médicament est propre à la MH : il s’agit de la tétrabénazine qui a
été développé pour contrôler la chorée (Frank 2014). Les autres traitements disponibles
actuellement, sont des antidépresseurs et anxiolytiques utilisés pour soigner les troubles
psychiatriques observés dans la pathologie. Il est également possible de mettre en place des
plannings ou de la rééducation pour les patients afin d’améliorer leur quotidien et de les
accompagner. Face à ce manque de thérapie ciblant efficacement la MH, de nombreuses
5molécules et traitements non médicamenteux font actuellement l’objet d’essais cliniques.
C’est le cas par exemple de médicaments testés pour traiter l’inflammation chronique
observée dans la pathologie (Denis, Lauruol, and Cicchetti 2019). Cependant, ces études
connaissent des résultats mitigés expliquant ainsi la poursuite des études précliniques dans
ce domaine. Par ailleurs, la stimulation cérébrale profonde, procédure chirurgicale
consistant à l’implantation d’un stimulateur électrique dans le cerveau dans le principal but
de contrôler les troubles moteurs, a été utilisée chez certains patients atteints de la MH
(Wojtecki et al. 2016). Les résultats observés chez ces patients sont plutôt positifs sur le
plan des symptômes moteurs, mais il ne s’agit pas encore d’une procédure couramment
utilisée dans le cadre de la prise en charge de la pathologie. Enfin, étant donné que la MH
résulte d’une mutation génétique, il a été envisagé d’agir directement sur la mutation par
l’utilisation dans un premier temps d’ARN (acide ribonucléique) interférent. Ainsi, un essai
clinique, IONIS-HTTRx (NCT02519036), a été mené chez 46 patients en phase 2a de 2015
à 2019. Les résultats observés indiquent une diminution de la présence de mHTT dans le
liquide céphalorachidien des patients ayant reçu le traitement (Tabrizi et al. 2018). Depuis
quelques années, une technique d’édition génomique, CRISPR/cas9 a été développée afin
de corriger de façon permanente une mutation. Des études précliniques utilisant cette
méthode sont en cours, et une équipe a déjà pu montrer qu’en corrigeant la mutation dans le
striatum, la thérapie permettait l’amélioration des symptômes moteurs et une diminution
significative du nombre d’agrégats dans la région traitée (S. Yang et al. 2017). Cependant,
l’effet n’a pas eu d’incidence sur la longévité des animaux.
Modèles animaux
Les souris sont de loin les animaux les plus couramment utilisés pour la modélisation de la
MH et ce depuis longtemps. Avant toutes les connaissances permettant la création de
modèles animaux transgéniques, les modèles animaux étaient crées par des lésions induites
par des neurotoxines de la région striatale dans le but de reproduire la perte de neurones
GABAergiques caractéristique de la MH (McGeer and McGeer 1976; Beal et al. 1986;
Schwarcz et al. 1984). Cependant, cette technique ne permettait pas de mimer fidèlement la
pathologie c’est pourquoi après l’apparition des modèles créés par modification génétique,
elle a été grandement mise de côté. Actuellement, plus de 25 modèles différents de souris
6de cette maladie ont été générés, dépendants de plusieurs approches à savoir : l’utilisation
de l’exon 1 uniquement ou de la totalité du gène HTT (knock-in) ; la longueur de la
répétition CAG, contenant ou non des codons CAA, incorporée dans la construction
génétique ; ou encore les différents types de promoteurs pour conduire l’expression de la
protéine mutée (Pouladi, Morton, and Hayden 2013). Ces différentes approches permettent
le développement de modèles qui miment plus ou moins bien la pathologie et de manière
plus ou moins rapide. Parmi ceux-ci, on peut prendre pour exemple le modèle R6/2. Cette
souris exprime l’exon 1 de la HTT humaine contenant une répétition instable de 144 CAG,
guidé par son promoteur humain. Ce modèle est très agressif ; en effet, la souris R6/2
développe la pathologie à partir de 8 semaines et présente de forts symptômes cognitifs et
moteurs (Mangiarini et al. 1996). Au contraire, il existe des modèles de souris chez lesquels
la pathologie sera plus lente et progressive (au delà de 6mois) ; c’est le cas des souris
zQ175 qui expriment tous les exons de la HTT humaine contenant une répétition d’environ
188 CAG ou encore des souris BACHD, qui expriment elles aussi tous les exons de la HTT
humaine contenant une répétition de 97 CAG stabilisé par la présence de codons CAA ; le
modèle BACHD est utilisé pour l’étude présentée au Chapitre 1 (Menalled et al. 2012;
Gray et al. 2008).
La protéine Huntingtine
Caractéristiques
Généralités
Le gène HTT, codant pour la HTT, contient 67 exons cependant, il peut subir un
épissage alternatif donnant alors différents variants de la protéine. Si on se place dans le cas
où la queue polyQ comporte 23 résidus glutamines, la HTT se compose de 3 144 acides
aminés et a un poids moléculaires de 348 kDa (Tartari et al. 2008). Dans le cas de la MH, la
mutation porte sur le nombre de répétition du triplet CAG, localisée dans l’exon 1 de HTT.
Il est important de souligner que le gène HTT est très conservé au cours de l’évolution ;
c’est à dire qu’on le retrouve chez les vertébrés comme chez les insectes. Cependant, l’exon
1 est la seule partie du gène à avoir varié. En effet, la présence d’une longue queue polyQ
7est spécifique aux mammifères, mais c’est chez l’humain que le nombre de CAG est le plus
important (Saudou and Humbert 2016).
L’exon 1 du gène HTT est divisé en trois domaines. Un premier domaine N-
terminal composé de 17 acides aminés très conservés chez les vertébrés (Tartari et al.
2008). Ce domaine consiste en une hélice α amphipathique importante pour la rétention
dans le réticulum endoplasmique (Atwal et al. 2007). Il agit comme un signal d’export
nucléaire et est sujet à des modifications post-traductionnelles influant sur la dégradation
et/ou la localisation des protéines (acétylation, sumoylation, ubiquitination et
phosphorylation) (Atwal et al. 2007; L. M. Thompson et al. 2009; Maiuri et al. 2013; J. S.
Steffan et al. 2004). Un deuxième, très polymorphique chez l’humain correspondant à la
queue polyQ entièrement composée de glutamines. Bien que ce domaine soit responsable
de la MH, son rôle reste inconnu. D’après la littérature, il pourrait être impliqué dans
l’autophagie. En effet, il a été montré que la suppression de ce domaine chez la Souris
entraine une augmentation de l’autophagie et de la longévité (Zheng et al. 2010). Enfin, un
troisième domaine composé de 50 acides aminés et enrichi en prolines (DRP), est retrouvé
uniquement chez les mammifères. Le DRP est lui aussi sujet à des polymorphismes dans la
population générale et est impliqué dans les interactions protéine-protéine. Malgré cette
fonction démontrée, sa délétion ne semble pas avoir d’effet néfaste chez la Souris
(Neveklovska et al. 2012). Pris ensemble, les domaines N-terminal et DRP sembleraient
jouer un rôle dans la capacité d’agrégation de la protéine HTT. Ainsi, ceci pourrait
expliquer les différences symptomatologiques observées chez des patients ayant le même
nombre de répétition CAG (Kuiper et al. 2017).
Le reste de la protéine est mal caractérisé. En effet, les 66 autres exons codant les
97.8% de la HTT sont très peu étudiés ; cela semble se justifier par le fait que la mutation
se situe dans l’exon 1. Dans cette partie, il est retrouvé de nombreuses répétitions HEAT
(protéine tandem repeat structural motif) : impliquées dans les interactions protéine-
protéine et retrouvées dans la HTT, la protéine phosphatase 2A, le facteur d’élongation 3 et
TOR1 (Palidwor et al. 2009). Les répétitions HEAT ont également un rôle dans les
interactions intramoléculaires permettant ainsi à la HTT d’adopter diverses formes
tridimensionnelles ; d’après la littérature, on en répertorie une centaine (Seong et al. 2010).
8Par ailleurs, on peut noter que dans l’exon 18, on retrouve une séquence d’exportation
nucléaire pouvant réguler la fonction ou la localisation de la HTT (Xia et al. 2003).
Figure 0.2 : Structure du gène de la huntingtine humaine. Abréviations : PolyGln :
queue polyglutamine; polyPro : domaine riche en proline ; HEAT : région de clivage
protéique ; NES : signal d’export nucléaire; NLS : signal de localisation nucléaire. D’après
(Déglon 2017).
Rôle de la HTT
La HTT est une protéine ubiquitaire retrouvée à différents niveaux à travers
l’organisme mais son expression est plus forte au sein du SNC (Marques Sousa and
Humbert 2013). Il s’agit d’une protéine essentielle ; en effet, son inhibition pendant le
développement embryonnaire est létal au jour 7.5 (Nasir et al. 1995; Duyao et al. 1995;
Zeitlin et al. 1995). De plus, des études axées sur l’embryogénèse ont montré qu’elle était
indispensable à la différentiation des neuroblastes dans le striatum, le cortex et le thalamus
(White et al. 1997; Reiner et al. 2001). Elle est aussi impliquée dans la migration des
neurones corticaux allant de la zone ventriculaire à la plaque corticale, dans l’homéostasie
cérébrale et la circulation du liquide cérébrospinal en agissant sur la biogénèse des cils des
épendymocytes, entrainant alors de possibles hydrocéphalies ou la fermeture de l’aqueduc
de Sylvius (Keryer et al. 2011; Dietrich et al. 2009). A contrario, il est important de noter
que son inhibition chez la Souris adulte entraine peu ou pas d’effets sur les fonctions
motrices ou cognitives et sur la longévité (Wang et al. 2016).
La HTT est impliquée dans divers mécanismes physiologiques. Elle est retrouvée en
grande partie dans le cytoplasme mais également dans le noyau. Son action intranucléaire
9est multiple : elle est impliquée dans la régulation de la transcription de nombreux facteurs
de transcription comme le NeuroD, le nuclear factor-kB (NF-kB) et la protéine 53
suppresseur de tumeur (p53). Elle agit également avec des activateurs ou répresseurs de la
transcription tels que le co-activateur TAFII130 ou le co-répresseur nucléaire (NCOR).
Enfin, elle interagit avec les récepteurs nucléaires incluant PPAR-γ, ou le récepteur à la
vitamine D et a une action sur la survie cellulaire à travers le blocage de l’activation des
caspases 3 et 9 prévenant ainsi l’apoptose (Rigamonti et al. 2001; Zhang et al. 2003), ou
bien en favorisant la transcription et le transport du BDNF (Brain-Derived Neurotrophic
Factor : facteur neurotrophique dérivé du cerveau)(Zuccato et al. 2001; Gauthier et al.
2004).
Dans le cytoplasme, elle agit sur le transport vésiculaire antérograde et rétrograde
par son interaction directe avec des protéines telles que la dynéine ou indirecte avec la
protéine associée à la huntingtine 1 via la dynactine et la kinésine (Caviston et al. 2007;
Gunawardena et al. 2003; S. H. Li et al. 1998; McGuire et al. 2006; Strehlow, Li, and
Myers 2019). Ainsi, la HTT régule le transport d’organelles incluant des vésicules
contenant la protéine VAMP7 v-SNARE (Colin et al. 2008), le BDNF (Gauthier et al.
2004), des vésicules précurseurs synaptiques (Zala, Hinckelmann, and Saudou 2013), ou
encore l’APP (amyloid precursor protein : protéine précurseur de l’amyloid)(Colin et al.
2008; Her and Goldstein 2008). De plus, à travers son action sur le transport antérograde,
elle joue un rôle important dans l’autophagie au niveau des axones ainsi que dans le
transport des protéines ubiquitinylées de l’autophagosome (Martin et al. 2014; Joan S.
Steffan 2010; Wong and Holzbaur 2014). On peut également noter son rôle dans la division
cellulaire en assurant l’orientation du fuseau mitotique (Elias et al. 2014; Godin et al. 2010;
Gutekunst et al. 1995), dans la ciliogenèse (Haremaki, Deglincerti, and Brivanlou 2015;
Keryer et al. 2011), dans l’endocytose (Engqvist-Goldstein et al. 2001; Legendre-Guillemin
et al. 2002; Waelter et al. 2001), le recyclage vésiculaire (Modregger et al. 2003) ou encore
le traffic endosomal (Pal et al. 2006).
10Figure 0.3 : Structure et transformation de la HTT. L'expression de HTT génère un
transcrit d'ARN initial qui est normalement transformé en un ARNm codant pour la HTT
(1), mais peut également être transformé de manière aberrante en un ARNm codant
uniquement l’exon 1 si le gène contient une expansion CAG (2). La traduction génère soit
la HTT (3), soit la protéine mHTT (4). Le clivage protéolytique médié par des séquences de
reconnaissance situées dans les segments désordonnés génère une série de produits, y
compris des fragments de type mHTT (5). Ces fragments contenant des queues polyQ
étendues jouent un rôle important dans le déclenchement de la MH. D’après (Bates et al.
2015)
La HTT mutée
Comme explicité précédemment, la MH résulte d’une répétition anormalement
longue de CAG. Celle-ci entraine alors la formation d’un ARN messager aberrant codant
seulement pour l’exon 1 du gène ce qui conduit alors à la synthèse de la partie N-terminal
de la protéine contenant la queue polyQ, considérée comme étant la principale forme
toxique de la mHTT (Sathasivam et al. 2019; Ross and Tabrizi 2011). D’autres formes de
HTT peuvent être également retrouvées par protéolyse de la protéine entière. En effet, la
HTT possède divers sites de clivages protéolytiques incluant des caspases, des cathépsines,
11des calpaines, ou encore des métalloprotéinases (Kim et al. 2001; Gafni and Ellerby 2002;
Hermel et al. 2004; Lunkes et al. 2002; Tebbenkamp et al. 2012). Ces observations
concernant la protéolyse ont été réalisées in vitro mais n’ont jamais été rapportées in vivo
(Y.P. Goldberg et al. 1996).
Cependant, c’est sous forme agrégée que l’on retrouve principalement la mHTT
dans la MH. En effet, en s’autoassemblant, elle donne de nombreuses conformations plus
ou moins toxiques. Ainsi, de nombreuses études ont été menées afin de déterminer la
pathogénicité des différentes formes et il semblerait que la mHTT sous forme fibrillaire
serait la plus toxique comparée à la forme agrégée (Drombosky et al. 2018; Hoop et al.
2016). L’agrégation de la mHTT est très semblable à l’agrégation protéique retrouvée dans
les maladies neurodégénératives comme la Maladie de Parkinson (MP) et la Maladie
d’Alzheimer (MA)(Fernàndez-Busquets 2013). Ainsi, dans ces pathologies on observe la
présence d’agrégats protéiques dans les tissus : l’α-synucléine pour la MP et la β-amyloïde
pour la MA. Ces protéines précurseurs de fibrilles ont une propension à former des feuillets
β hautement importants pour les interactions protéines-protéines. Dans le cas de la mHTT,
c’est la formation d’épingles à cheveux β qui initie l’agrégation (Hoop et al. 2016). Nota
bene, il y a une corrélation positive directe entre la taille de la queue polyQ et la formation
de ces structures entrainant l’agrégation (Kar et al. 2011).
Tout comme la HTT, la mHTT est elle aussi sujet à des clivages par des protéases
formant ainsi des fragments N-terminaux contenant la queue polyQ anormale ; fragment le
plus toxique de la HTT (Ross and Tabrizi 2011). Une corrélation inverse est établie entre la
taille de ce fragment et sa toxicité : plus le fragment est court et plus sa toxicité est forte.
Son pouvoir pathogène résulterait de sa capacité à transloquer dans le noyau et à perturber
la transcription induisant alors la mort cellulaire (Saudou et al. 1998). De plus, pour
compliquer quelque peu la situation, la mHTT peut inhiber le protéasome et l’autophagie,
entrainer des dysfonctionnements mitochondriaux et du transport microtubulaire ou encore
altérer l’endocytose, l’activité synaptique et promouvoir l’excitotoxicité (Ross and Tabrizi
2011).
12Figure 0.4: Modèle schématique illustrant le mauvais repliement et l'agrégation de
protéines contenant une queue polyQ étendue. D’après (Adegbuyiro et al. 2017)
La protéine prion
Etant donné que les études in vitro et in vivo qui vont être présentées ensuite
évoqueront la théorie dite du « prion » concernant la pathogénicité de la mHTT, il est
d’abord nécessaire de présenter la théorie des maladies à prion établie sur la protéine
pathogénique prion.
Les maladies à prions sont des encéphalopathies spongiformes transmissibles
considérées également comme des maladies neurodégénératives infectieuses. On retrouve
parmi celles-ci la tremblante du mouton, l’encéphalopathie spongiforme bovine (« vache
folle ») ou encore la maladie de Creutzfeldt-Jakob chez l’Homme. En 1982, Prusiner
découvre la protéine prion cytoplasmique (PrPc), une protéine ubiquitaire chez l’Homme,
responsable de la pathologie de Creutzfeldt-Jakob (Prusiner 1982). Après infection, cette
protéine va adopter une conformation riche en feuillets β (PrP scrapie : PrPsc) la rendant
pathologique. Le changement de conformation évolue alors vers une forme amyloïde à
l’origine de vacuolisation neuronale entrainant la mort cellulaire. La protéine PrPsc acquiert
également des propriétés de propagation, lui conférant la capacité d’infecter de nouvelles
cellules en transmettant l’information pathogénique à la protéine saine contenu par un
phénomène de transconformation et d’autoréplication. La PrPsc a une capacité de
13transmission inter-individu, comme ceci a été montré par le phénomène des contaminations
par injections d’hormones de croissance provenant de l’hypophyse de patients atteints de la
maladie de Creutzfeldt-Jakob ; mais également inter-espèce, comme l’a expressément
montré l’épidémie de la « vache folle » dans les années 1960 par la contamination chez
l’Homme après ingestion de viandes bovines contaminées (Colby and Prusiner 2011).
Ainsi, la définition de prion pour une protéine requiert quatre caractéristiques : 1) le
changement conformationnel de la protéine native vers une forme pathogénique riche en
feuillets β capable de s’agréger, 2) la propagation intercellulaire capable d’infecter de
nouvelles cellules et de nouvelles structures, 3) la transmission de l’information
pathogénique à la forme native de la cellule hôte nouvellement infectée par
transconformation et enfin 4) la transmission inter-individuelle intra- ou inter-espèce.
Propagation de la mHTT
In vitro
Les premières études montrant les capacités de propagation de la mHTT ont été
réalisées in vitro. Ainsi, c’est en 2002 qu’une équipe a pu mettre en évidence que des
cellules (Cos-7 et PC12) étaient capables d’internaliser des agrégats de mHTT constitués de
peptides polyQ42, depuis l’espace extracellulaire (W. Yang et al. 2002). La toxicité des
peptides polyQ42 a été montrée lors de l’ajout d’une séquence de localisation nucléaire.
Une autre étude s’est intéressée à des fibrilles synthétiques polyQ44 de mHTT et a ainsi
montré leur capacité à s’incorporer dans le milieu intracellulaire en culture avec différents
types de cellules (Cos-7, HEK293, neuro2A, CHO et HeLa). Il semblerait que les fibrilles
traversent la membrane plasmique via un passage passif pour se retrouver dans le
cytoplasme, colocalisées parfois avec différentes protéines (Hsp70, ubiquitine, etc…). Par
ailleurs, il a été montré, en taguant la HTT avec une queue polyQ25 non pathologique dans
des cellules HEK293, que les fibrilles étaient capables d’entrainer l’agrégation de la HTT
endogène (Ren et al. 2009). De plus, ils ont montré que des cellules exprimant des fibrilles
de mHTT-Q71 étaient capables de se propager vers les cellules exprimant la forme non
mutée de la HTT et ainsi induire son agrégation. D’autres études ont ensuite été menées ;
elles ont confirmé ces premiers résultats in vitro mais ont permis d’y ajouter des notions de
14Vous pouvez aussi lire

















































