Phylogéographie du mélèze laricin (Larix laricina Du roi K. Koch) en Amérique du Nord - Mémoire Emile Warren Maîtrise en sciences forestières
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Phylogéographie du mélèze laricin (Larix laricina
[Du roi] K. Koch) en Amérique du Nord
Mémoire
Emile Warren
Maîtrise en sciences forestières
Maître ès sciences (M. Sc.)
Québec, Canada
© Emile Warren, 2015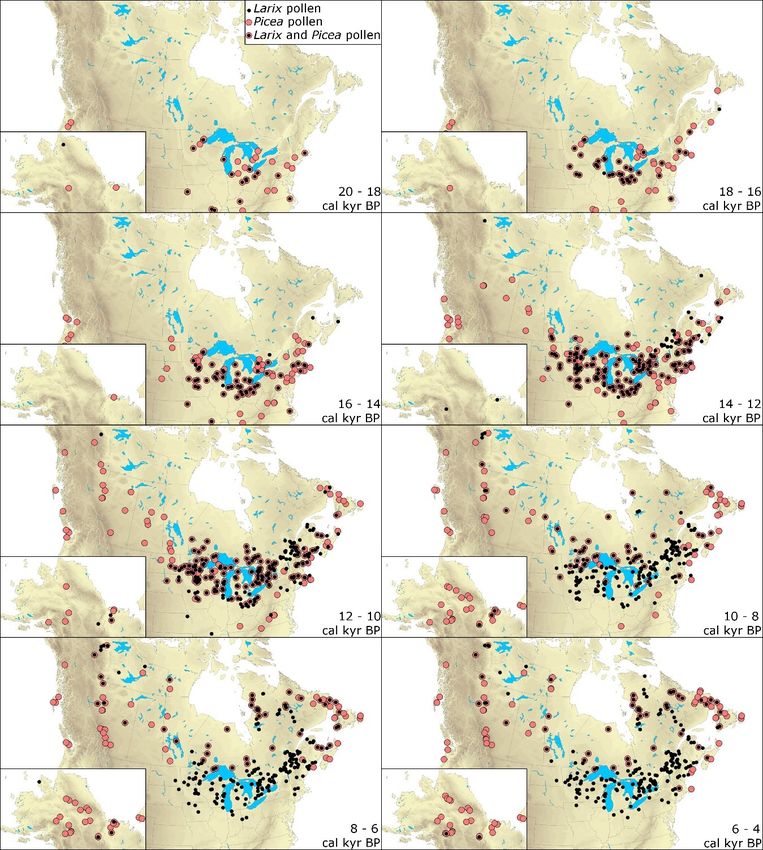
Résumé
La structure des populations du mélèze laricin (Larix laricina [Du roi] K. Koch) a été
étudiée à l’aide de polymorphismes de l’ADN mitochondrial et chloroplastique. Deux
populations, situées en Alaska et au Labrador, étaient génétiquement distinctes des autres,
suggérant l'existence de refuges glaciaires nordiques à ces endroits. La répartition spatiale
des haplotypes a révélé un clivage génétique entre deux groupes de populations occupant
l’est et l’ouest de l'aire de répartition. Ce patron témoignerait de la présence de deux lignées
glaciaires génétiquement distinctes provenant d’autant de refuges localisés au sud de
l'inlandsis Laurentidien. L’analyse des données polliniques a permis de corroborer la
présence de refuges glaciaires au sud-ouest des Grands Lacs et à l’ouest des Appalaches, en
plus des possibles refuges en Alaska et au Labrador. La haute différenciation génétique
propre aux populations de l’ouest pourrait être la conséquence d’une forte compétition
interspécifique lors de la recolonisation postglaciaire de cette région.
III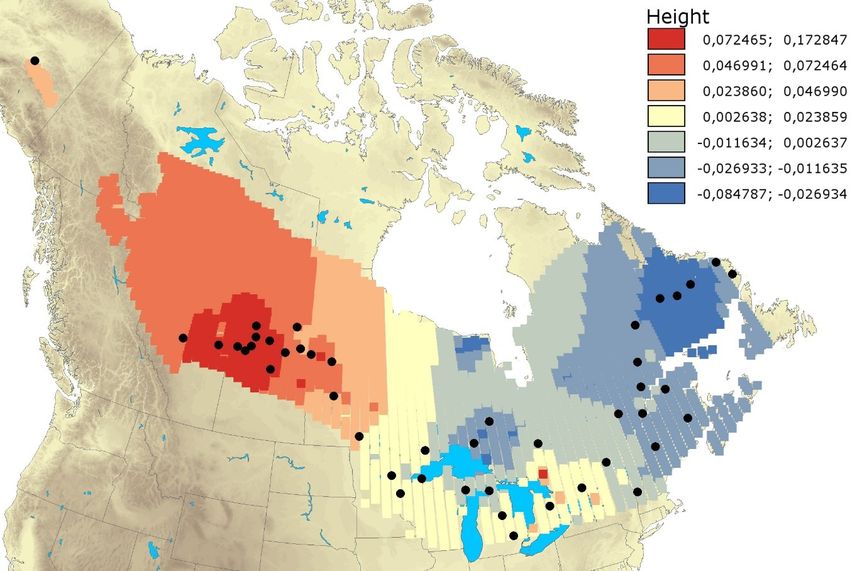
Abstract
Geographical population structure of the North American larch, Larix laricina [Du
roi] K. Koch was studied using mitochondrial and chloroplast DNA polymorphisms. Some
populations from Alaska and Labrador were genetically differentiated from neighboring
populations, suggesting that these two regions served as glacial refugia. The spatial
distribution of haplotypes revealed a cleavage between eastern and western populations,
which are probably representative of two distinct glacial lineages that expanded from the
south of the ice sheet following the last glacial maximum. Mapped pollen records helped
inferring the putative location of glacial refugia south-west of the Great Lakes, west of the
Appalachians, as well as in Alaska and Labrador. High population differentiation among
western populations likely indicates that interspecific competition was strong during the
postglacial colonization of the region.
V
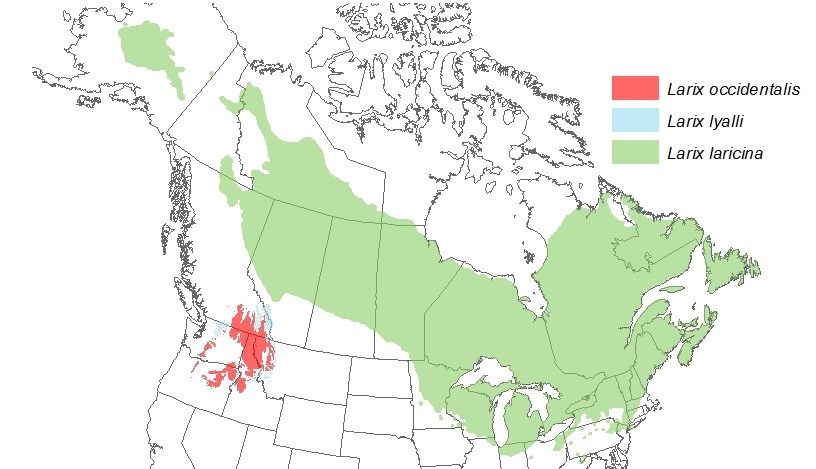
Table des matières
Résumé................................................................................................................................. III
Abstract ................................................................................................................................. V
Table des matières ..............................................................................................................VII
Liste des tableaux................................................................................................................. IX
Liste des figures ................................................................................................................... XI
Remerciements.................................................................................................................. XIII
Avant-propos ..................................................................................................................... XV
Chapitre 1 - Introduction générale ......................................................................................... 1
1.1 Phylogéographie ........................................................................................................... 1
1.1.1 Définitions et concepts .......................................................................................... 1
1.1.2 La dernière glaciation ............................................................................................ 2
1.1.3 Refuges glaciaires .................................................................................................. 3
1.1.4 Pollen et graines chez les conifères ....................................................................... 4
1.1.5 Données fossiles .................................................................................................... 5
1.2 Diversité génétique....................................................................................................... 5
1.2.1 Mécanismes affectant la diversité génétique ......................................................... 6
1.2.2 Propriétés génétiques des refuges glaciaires ......................................................... 7
1.2.3 Marqueurs génétiques ............................................................................................ 8
1.2.4 Analyse des données génétiques ........................................................................... 8
1.3 Les génomes cytoplasmiques des conifères ................................................................. 9
1.3.1 Génome mitochondrial .......................................................................................... 9
1.3.2 Génome chloroplastique ...................................................................................... 10
1.4 Espèce à l’étude : le mélèze laricin ............................................................................ 10
1.4.1 Le genre Larix ..................................................................................................... 10
1.4.2 Larix laricina ....................................................................................................... 11
1.4.2.1 Éco-physiologie de l’espèce ........................................................................ 12
1.4.2.2 Diversité génétique ...................................................................................... 13
1.4.2.3 Hybridation .................................................................................................. 14
1.5 Objectifs et hypothèses .............................................................................................. 14
1.6 Littérature citée .......................................................................................................... 16
Chapitre 2 – Histoire postglaciaire du mélèze laricin .......................................................... 23
2.1 Résumé ....................................................................................................................... 24
2.2 Abstract ...................................................................................................................... 25
2.3 Introduction ................................................................................................................ 26
2.4 Materials and methods ............................................................................................... 30
2.4.1 Sampling and DNA Extraction ............................................................................ 30
2.4.2 MtDNA polymorphism screening, PCR conditions and genotyping .................. 31
2.4.3 CpDNA polymorphism screening, PCR conditions and genotyping .................. 31
2.4.4 Genetic diversity, population structure and differentiation ................................. 32
2.4.5 Analysis of published pollen paleorecords .......................................................... 33
2.5 Results ........................................................................................................................ 34
2.5.1 Genetic diversity .................................................................................................. 34
2.5.2 Population structure and differentiation .............................................................. 36
2.5.3 Analysis of published pollen paleorecords .......................................................... 36
VII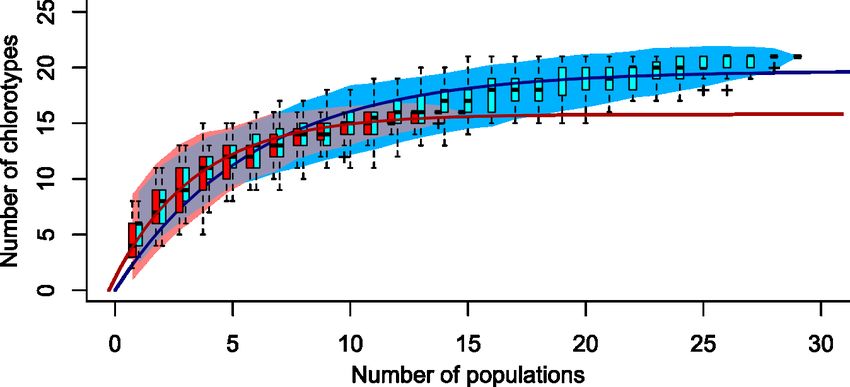
2.6 Discussion ................................................................................................................... 41
2.6.1 Genetic diversity and population differentiation ................................................. 41
2.6.2 Population structure and phylogeographic inferences .........................................42
2.6.3 High cpDNA population differentiation and competition in western Canada .....43
2.7 Conclusions ................................................................................................................ 45
2.8 Acknowledgements ....................................................................................................46
2.9 Literature cited ............................................................................................................ 47
Chapitre 3 - Conclusions et perspectives de recherche ........................................................ 51
3.1 Retour sur les objectifs et hypothèses de recherche ................................................... 51
3.2 Inférences phylogéographiques .................................................................................. 52
3.3 Différenciation génétique des populations de l’ouest ................................................. 54
3.4 Littérature citée ...........................................................................................................56
Liste entière de la littérature citée ........................................................................................ 59
Annexes ................................................................................................................................ 69
VIIIListe des tableaux
Table 2.1. Population genetic diversity estimates and Bayesian group assignment based on
three cpSSRs markers for 45 Larix laricina populations......................................... 38
Appendix S1. Mitotype counts for one mtDNA polymorphic locus in 45 Larix laricina
populations. ............................................................................................................. 69
Appendix S2. Chlorotype counts for three cpDNA polymorphic SSRs in 45 Larix laricina
populations. ......................................................................................................... 70-72
Appendix S3. List of mtDNA loci scanned for Larix laricina mtDNA polymorphism, primer
sequences, annealing temperature and sequencing results................................. 73-75
IXListe des figures
Figure 1.1. Simulation de l’aire recouverte par la calotte glaciaire nord-américaine lors du
dernier maximum glaciaire il y a 21 000 ans (i.e. 18 000 14C BP). La zone blanche
représente les territoires recouverts par la glace et la zone grise les territoires non
recouverts. Tiré de Dyke et al. 2004. ......................................................................... 3
Figure 1.2. Carte représentant l’aire de répartition des trois espèces du genre Larix en
Amérique du Nord. L’aire de répartition est montrée en vert pour Larix laricina, en
bleu pour Larix lyallii et en rouge pour Larix occidentalis...................................... 12
Figure 2.1. Biogeographic model illustrating contrasted scenarios of postglacial
recolonization by early-successional and late-sucessional species. Large ellipses
represent a modeled terrestrial landscape recently deglaciated (barren landscape;
white), colonized by early-successional species (yellow), or colonized by late-
successional species (green). The four small circles represent unproductive sites
where more extreme ecological conditions with respect to the surrounding landscape
prevail. These sites are either virtually treeless (white circles) or colonized by early-
successional, low resource-demanding species (yellow circles) but late-successional,
high resource-demanding, species cannot establish there. Both scenarios begin with a
recently deglaciated barren landscape and end with the current landscape dominated
by late-successional species interspaced by small, disjunct populations of the early-
successional species. The difference between the two scenarios is the chronology of
postglacial recolonization with the recently deglaciated landscape initially colonized
by an early-successional (A) or a late-successional (B) species. ............................. 29
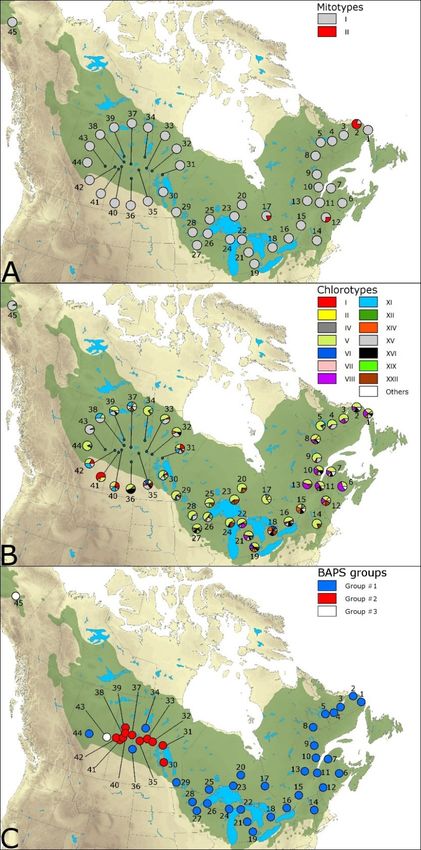
Figure 2.2. Geographic distribution of mitotypes (A), chlorotypes (B), and (C) Bayesian
clustering of 45 Larix laricina populations. Chlorotypes with a frequency below 0.01
were pooled as “Others”. Bayesian clustering based on chlorotype frequencies using
the “spatial clustering of groups” option implemented in BAPS 6.0 (Corander et al.,
2008). Population numbers correspond to those in Table 2.1. ................................. 35
Figure 2.3. Among-population relative genetic distances using cpDNA data across Larix
laricina natural range. The analysis was carried out with the Alleles in Space (AIS)
software using the “genetic landscape shapes” procedure (Miller, 2005). The “height”
value represents population differentiation at a given point. The higher the “height”,
the higher the relative population differentiation. Interpolation performed with a grid
of 100 × 100 and a distance weighting parameter of 1 (see Miller et al., 2006 for
details). ..................................................................................................................... 37
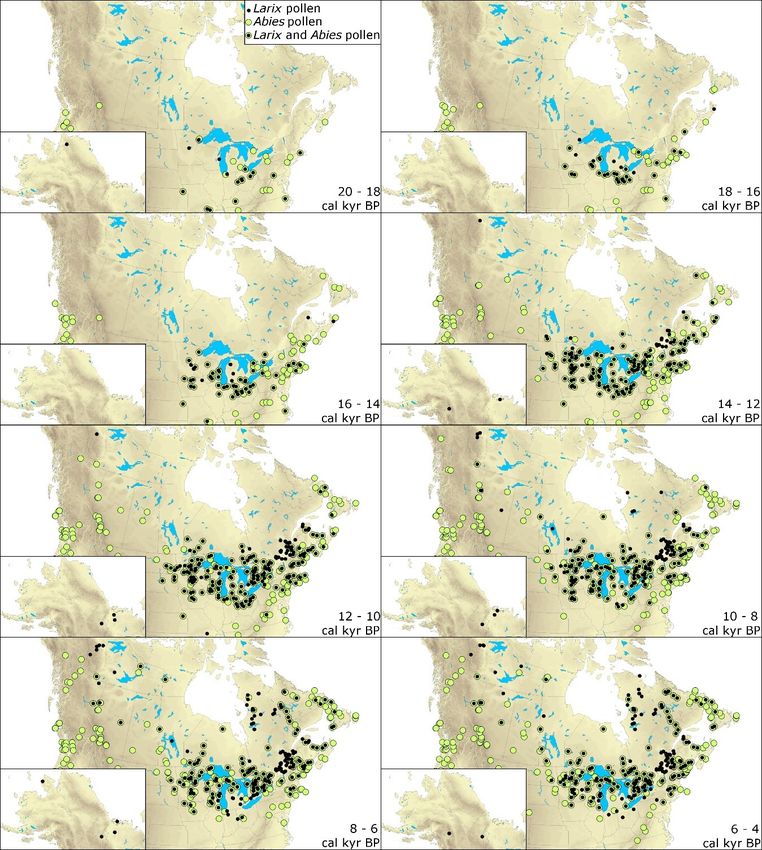
Figure 2.4. Mapped pollen paleorecords of Larix sp. and Picea sp. between 20 and 4
calibrated kiloyears before present (cal kyr BP) reconstructed from 408 and 466
pollen cores, respectively. Black and pink dots indicate the presence of larch and
spruce pollen in the paleorecord during each 2 kyr time-interval, respectively. ..... 39
Figure 2.5. Mapped pollen paleorecords of Larix sp. and Abies sp. between 20 and 4
calibrated kiloyears before present (cal kyr BP) reconstructed from 408 and 490
pollen cores, respectively. Black and green dots indicate the presence of larch and fir
pollen in the paleorecord during each 2 kyr time-interval, respectively. ................. 40
XIFigure S1. Accumulation curves of the number of chlorotypes uncovered as a function of
the number of populations sampled. Western and Eastern lineages are represented by
red and blue colors, respectively. Each polygon represents 95% confidence interval
envelopes of 100 randomly permuted accumulation curves. Boxplots of the 100
permutations indicate lower quartile, median and upper quartile; whiskers length are
1.5 × interquartile ranges. Filled lines are the extrapolation curves fitted from each
mean accumulation curve using an asymptotic, negative exponential function. .....79
XIIRemerciements
J’aimerais d’abord remercier Jean Bousquet qui m’a donné l’opportunité de réaliser
ce projet et m’a fourni toutes les ressources pour accomplir des avancées en phylogéographie,
discipline qui m’était complètement étrangère. Merci de m’avoir permis de mener ce projet
à terme et d’acquérir autant d’expérience. J’aimerais aussi remercier Jean Beaulieu du Centre
de foresterie des Laurentides et Martin Perron du Ministère des forêts, de la faune et des parcs
du Québec pour m’avoir fourni des échantillons ou de l’aide lors de l’échantillonnage. Un
merci également à Julie Godbout et Benjamin Cinget pour avoir échantillonné certaines
populations éloignées de mélèze laricin et à Juan-Pablo Jaramillo-Correa pour avoir conçu la
seule amorce mitochondriale ayant révélé des résultats pertinents dans le cadre de mon étude.
J’aimerais également remercier toute l’équipe de professionnels de recherche et
d’étudiants-chercheurs du laboratoire. Sauphie Senneville, qui m’a fourni une aide
incalculable pendant les premières années du projet, autant comme « coach » en ce qui
concerne les méthodes qu’en fournissant un temps précieux pour m’aider à effectuer les
innombrables amplifications PCR avant de partir en congé de maternité. Sébastien Gérardi,
pour toutes ces discussions sur la génétique et les notions qui n’étaient pas toujours claires.
Merci infiniment, collègue, pour toute ton aide dans les analyses, la rédaction et le reste.
Guillaume de Lafontaine, pour ses talents d’homme de pollen, merci de t’être infiltré dans la
foule avant la ligne d’arrivée pour ajouter à ce travail la touche qui lui manquait. Merci à
Sylvie Blais et France Gagnon pour leur « coaching » dans le laboratoire et pour m’avoir
enseigné une multitude de techniques, et à Marcel Laurent Estrada et Francis Tremblay pour
leur aide dans les débuts avec les millions de manipulations à faire.
Merci aussi à tous mes camarades de bureau et du deuxième étage de l’Institut de
biologie intégrative et des systèmes au pavillon Marchand. Mebarek Lamara et Juliana Sena,
impossible d’imaginer de meilleurs comparses de bureau, vous êtes les prochains à déposer
vos thèses, tenez bon; Gaby Germanos, Julien Prunier, Mathieu Paradis, pour ces dizaines de
conversation de coin de corridor ou de rackage de tips.
XIIIFinalement, merci à ma copine Marie-Pier de m’avoir épaulé et enduré pendant ces trois dernières années de travail insensées à aller au lit à 5 heures du matin. Merci, on est partis pour la gloire! XIV
Avant-propos
Le premier chapitre de ce mémoire est une introduction générale comprenant une
revue de littérature qui traite des principaux concepts nécessaires à la compréhension des
thèmes abordés dans le reste du mémoire. Ce chapitre se conclut sur la présentation des
objectifs et hypothèses qui ont guidé le travail effectué.
Le deuxième chapitre est un article scientifique rédigé en anglais par l’étudiant et
corrigé par les co-auteurs. Jean Bousquet (directeur) et Jean Beaulieu (codirecteur) ont
participé à la conception du projet, Sauphie Senneville a aidé à réaliser les travaux
expérimentaux en laboratoire, Martin Perron et Jean Beaulieu ont fourni de l’aide à
l’échantillonnage, Juan-Pablo Jaramillo-Correa a conçu les amorces mitochondriales et
Sébastien Gérardi et Guillaume de Lafontaine ont fourni de l’aide avec les analyses. Tous
ont participé à la rédaction du manuscrit. L’article est destiné à être publié dans une revue
scientifique internationale. Il est lui-même composé de cinq sous-parties. La première partie
est une introduction synthétique reprenant les concepts essentiels de l’introduction générale
du mémoire. La deuxième partie, le matériel et les méthodes, contient l’information
nécessaire pour reproduire l’étude ainsi qu’une explication détaillée des analyses effectuées.
La partie suivante est composée des résultats obtenus qui sont interprétés et analysés dans
l’avant-dernière partie, où l’on discute des implications de ces derniers. L’article se termine
par une conclusion.
Le troisième chapitre est une conclusion générale du mémoire. Cette partie contient
les conclusions quant à l’atteinte des objectifs de l’étude et la vérification des hypothèses
posées lors de l’introduction du mémoire. Elle se termine par un récapitulatif des limites de
l’étude et des perspectives de recherche futures permettant de répondre à ces limites et
d’étendre la portée de l’étude.
XVChapitre 1 - Introduction générale
1.1 Phylogéographie
1.1.1 Définitions et concepts
La biogéographie est l’étude de la répartition géographique des espèces (MacArthur,
1967). Elle cherche à décrire la répartition et l’abondance des individus ou des populations à
différentes échelles spatiales en considérant les facteurs historiques, géologiques,
géographiques et biotiques (Humphries, 2000).
La phylogénie est l’étude des relations entre les espèces actuelles et leurs ancêtres.
Elle a pour but d’établir des liens de parenté entre les espèces et de retracer leur histoire
évolutive à travers le temps. Les informations utilisées pour construire un arbre
phylogénétique proviennent traditionnellement de traits morphologiques et d’outils
moléculaires (Bousquet et al., 1992). La phylogénie peut également s’intéresser aux liens de
parenté entre les individus ou les populations d’une même espèce, et donc s’appliquer à
différents niveaux taxonomiques.
La phylogéographie peut être considérée comme l’union de la phylogénie et de la
biogéographie. Elle consiste à projeter un arbre phylogénétique à l’échelle d’une espèce sur
un support géographique (Avise, 2000). Le but est de retracer l’histoire des différentes
populations à travers le temps et l’espace pour ensuite inférer les événements climatiques ou
géologiques passés ayant causé leur répartition actuelle (Bermingham & Moritz, 1998;
Hewitt, 2000; Soltis et al., 2006; Jaramillo-Correa et al., 2009). Les oscillations climatiques
et les phénomènes de vicariance (barrières physiques ou isolement géographique séparant
deux populations anciennement unies) peuvent causer des bases d’échanges génétiques (flux
génique), des goulots d’étranglement (réduction de la taille effective d’une population
entraînant une réduction de sa diversité génétique et la dérive aléatoire de cette dernière) ou
encore, l’expansion des populations, pouvant entraîner des divergences dans la composition
génétique des populations (Arbogast & Kenagy, 2001).
La phylogéographie nécessite donc d’étudier un ou plusieurs traits ayant évolué dans
le temps tout en laissant des vestiges ou « signatures », dans les populations contemporaines
1de l’espèce étudiée. Pour ce faire, l’outil utilisé le plus fréquemment dans les études récentes
est le polymorphisme génétique, soit la variation du code génétique des individus. En effet,
l’ADN (Acide désoxyribonucléique) évolue dans le temps à travers ses mécanismes de
réplication qui modifient progressivement les séquences nucléotidiques au fil des réplications
(mutations). Lorsque ces changements sont conservés, ils laissent des traces dans les
populations actuelles. Si ces polymorphismes sont structurés géographiquement, c’est-à-dire
répartis de façon non aléatoire dans l'espace, il est possible de retracer l’histoire
phylogéographique de l’espèce (Avise et al., 1987). Lorsque l’on compare plusieurs études
phylogéographiques réalisées sur un même territoire, il est même possible d’observer des
tendances multi-espèces et d’inférer les processus historiques communs qui ont modelé leur
répartition géographique actuelle, augmentant la puissance d’inférence de l’approche
(Bermingham & Moritz, 1998; Jaramillo-Correa et al., 2009).
1.1.2 La dernière glaciation
Le début du dernier événement glaciaire majeur remonte à environ 135 000 ans. Le
dernier maximum glaciaire (Last Glacial Maximum; LGM) est quant à lui décrit comme le
moment où un maximum d’eau était séquestré par les glaces à l’échelle de la planète (Mix et
al., 2001). Cela correspond à la période où le niveau d’eau eustatique était à son plus bas. Il
aurait été atteint entre 23 000 et 19 000 cal. yr. BP. (Mix et al., 2001).
En Amérique du Nord, la glace aurait recouvert progressivement les régions
nordiques jusqu’à ce que les plaques glaciaires recouvrent le Canada presqu’entièrement.
Durant le LGM, la calotte glaciaire s’étendait sur les dix provinces et trois territoires
canadiens, ainsi que sur une partie du nord des États-Unis (Figure 1.1) (Dyke et al., 2002).
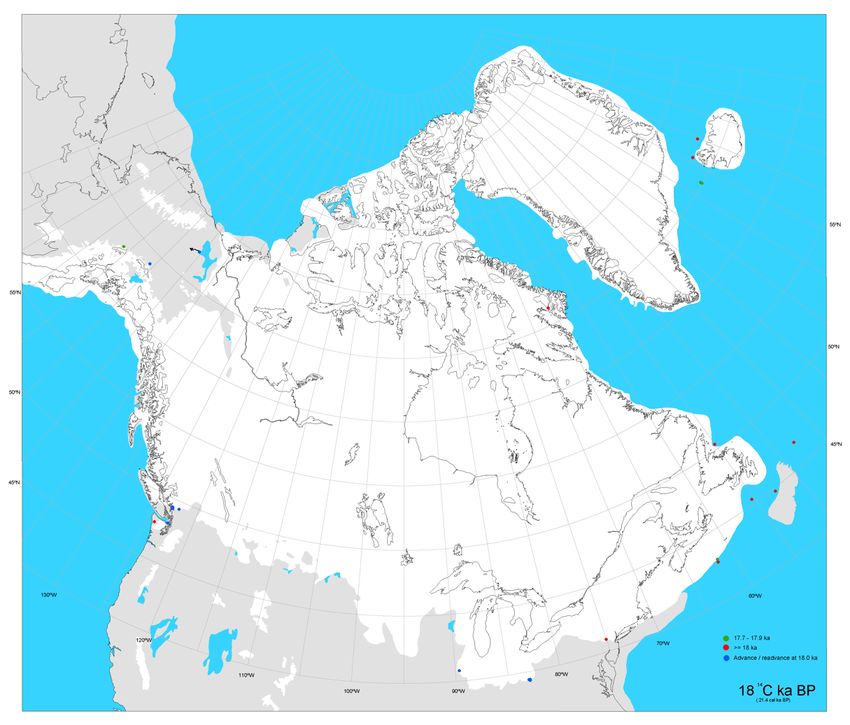
2Figure 1.1. Simulation de l’aire recouverte par la calotte glaciaire nord-américaine lors du
dernier maximum glaciaire il y a 21 000 ans (i.e. 18 000 14C BP). La zone blanche représente
les territoires recouverts par la glace et la zone grise les territoires non recouverts. Tiré de
Dyke et al. 2004.
1.1.3 Refuges glaciaires
Lors des périodes glaciaires du Quaternaire, les espèces ont vu leur environnement
changer énormément (Webb & Bartlein, 1992; Davis & Shaw, 2001). La vie macroscopique
dans les zones terrestres recouvertes de glace devenant très difficile voire impossible, la
progression des glaces aurait repoussé les différentes espèces vers les régions méridionales
libres de glace. De tels endroits où les espèces ont pu survivre aux époques glaciaires et ainsi
éviter l’extinction (Jackson et al., 1997) sont appelés refuges glaciaires (Shafer et al., 2010;
Keppel et al., 2012; Gavin et al., 2014).
En Amérique du Nord, les refuges glaciaires présumés les plus couramment évoqués
étaient situés au sud des glaciers. Du côté oriental du continent, la chaîne de montagnes des
3Appalaches sépare deux refuges principaux qui auraient été situés à l’est des Appalaches et
au sud des Grands Lacs (Godbout et al., 2005; Jaramillo-Correa et al., 2009; Gérardi et al.,
2010). À l'ouest du continent, plusieurs refuges ont été identifiés dont un qui aurait été
localisé à l’ouest des Rocheuses, la chaîne de montagnes agissant comme barrière physique
à la migration (Jaramillo-Correa et al., 2004; Godbout et al., 2008; Wei et al., 2011).
Certaines études mentionnent la possibilité d’un refuge sur le plateau continental du Labrador
exondé au LGM (Jaramillo-Correa et al., 2004; Gérardi et al., 2010) ainsi qu’en Alaska
(Béringie) (Hopkins, 1982; Brubaker et al., 2005; Anderson et al., 2006; Zazula et al., 2006;
de Lafontaine et al., 2010; Gérardi et al., 2010). Cette dernière région était libre de glace au
LGM et semble donc avoir été un refuge de prédilection pour plusieurs espèces ayant une
aire de répartition nordique (Jackson et al., 2000). En plus de ceux-ci, certains auteurs
mentionnent l'existence possible de micro-refuges glaciaires cryptiques localisés à la marge
de la calotte glaciaire dont on ignore l’emplacement exact puisque certains pourraient
aujourd’hui être localisés sur le plateau océanique continental maintenant inondé (Tremblay
& Schoen, 1999; McLachlan et al., 2005; Godbout et al., 2010; Shafer et al., 2010). Ces
derniers ont été suggérés pour expliquer la présence hâtive de certaines espèces à des endroits
jugés inatteignables dans la fenêtre de temps donnée selon leur vitesse de migration
(McLachlan et al., 2005). À la suite du retrait des glaces, les espèces ont pu progressivement
recoloniser les terres nouvellement déglacées à partir des divers refuges glaciaires pour
aboutir à la répartition actuelle des populations (Pielou, 1991).
1.1.4 Pollen et graines chez les conifères
Le flux génique des génomes des organelles dans la famille des Pinaceae est transmis
de façon bilatérale et uniparentale, soit par la mère (matrilinéaire) ou par le père
(patrilinéaire). Le pollen, gamétophyte mâle, transmet le génome chloroplastique (ADNcp)
(Neale & Sederoff, 1988) et l’ovule, gamète femelle, transmet le génome mitochondrial
(ADNmt) (Dong & Wagner, 1993).
Cela donne lieu à des patrons de dissémination distincts selon le génome étudié,
puisque le flux génique par graine ou par pollen est différent chez les Pinaceae (Petit et al.,
2005; Jaramillo-Correa et al., 2009). Le pollen, plus léger, peut être transporté par le vent et
être disséminé sur de longues distances, tandis que la graine, plus lourde, voyage moins
4aisément. On la retrouvera plutôt dans un périmètre proximal de la mère (Petit & Vendramin,
2007). De manière générale chez les Pinaceae, on admet que l’ADNmt a une capacité de
dissémination plus faible que l’ADNcp puisque la graine parcourt une distance plus courte
que le pollen. Par conséquent, les barrières physiques limitant leur dissémination agissent
différemment et on s’attend à obtenir une meilleure dispersion de l’ADNcp et une plus grande
uniformité géographique de la structure génétique des populations. En phylogéographie, on
cherchera cependant à obtenir la plus grande résolution possible des structures géographiques
anciennes et une différenciation génétique maximale des populations, l’ADNmt sera alors le
génome de choix (Avise et al., 1987; Jaramillo-Correa et al., 2009).
1.1.5 Données fossiles
Dans un contexte d’étude de l’histoire biogéographique d’une espèce, les données
fossiles de pollen et de macrofossiles peuvent également être comparées aux résultats des
analyses de structure génétique de populations. Le registre fossile permet de confirmer la
présence d’une espèce ou d’un genre (s’il n’est pas possible de discriminer
morphologiquement le pollen d’espèces d’un même genre) à un endroit et un moment donné.
Néanmoins, on peut inférer la localisation des refuges glaciaires potentiels en s’appuyant
simultanément sur les données moléculaires, les simulations d’expansion des calottes
glaciaires telles que proposées par Dyke 2004 (Figure 1.1) et Marshall 2002 pour l’Amérique
du Nord ainsi que les relevés palynologiques et de macrofossiles (Gavin et al., 2014).
L'absence de pollen ou de macrofossiles à une date donnée n’est cependant pas suffisante
pour conclure avec certitude que l’espèce ou le genre n’était pas présent (Davis et al., 1991;
Birks, 2003). C’est pourquoi les inférences phylogéographiques intégratives à partir des
données génétiques et fossiles deviennent de plus en plus incontournables (Jaramillo-Correa
et al., 2009).
1.2 Diversité génétique
La diversité génétique est le degré de variation génétique à l’intérieur d’un taxon
donné. Toute différence dans la séquence d’ADN des individus à l’intérieur de ce taxon est
un polymorphisme et sert d’unité de base à la diversité génétique. Un locus (région spécifique
du génome, ex. : un gène) pour lequel on ne retrouve aucune variation génétique est dit
5monomorphe, tandis qu’un locus retrouvé sous plusieurs formes est considéré polymorphe.
Il existe quatre processus évolutifs qui agissent sur la diversité génétique et l’abondance de
polymorphismes : la mutation, la sélection naturelle, la dérive génétique et la migration.
1.2.1 Mécanismes affectant la diversité génétique
La mutation est le processus à l'origine de toute variation génétique. Elle agit
directement sur l'ADN des individus en modifiant leur séquence nucléotidique. Les
mécanismes de réplication de l’ADN n’étant pas infaillibles, chaque erreur de réplication
introduite crée un variant génétique du brin copié. Ces mutations peuvent être éliminées par
la sélection naturelle, mais elles sont parfois transmises à la prochaine génération (Thompson
et al., 1998). La progéniture possédera alors un code génétique unique et différent de celui
de ses parents.
La sélection naturelle est une force évolutive favorisant les polymorphismes qui
confèrent un phénotype avantageux à l’individu qui les porte. L’étude des polymorphismes
sous sélection ne présente pas d’intérêt en phylogéographie car ils reflètent des processus
récents (i.e. adaptation à un milieu). Par opposition, les marqueurs de choix en
phylogéographie sont dits neutres et ne reflètent que des processus historiques découlant de
façons évolutives particulières (Avise, 2000).
La dérive génétique résulte simplement de l'échantillonnage aléatoire des allèles d'une
population qui seront transmis à la nouvelle génération. Il s'agit donc d'un processus évolutif
neutre qui modifie aléatoirement la fréquence des allèles des populations au fil des
générations. Ce processus a un effet plus important dans les populations isolées avec une
petite taille efficace où le flux génique avec les autres populations est limité.
L'échantillonnage aléatoire des allèles qui intervient à chaque génération confère aux allèles
moins fréquents une chance statistiquement plus faible d’être transmis. On observe
généralement dans ces populations une diminution de la fréquence des allèles rares et, si
l’isolement persiste, leur disparition. On associe à la dérive une perte de diversité génétique
ainsi qu’une différenciation accrue entre les populations (Wright, 1932; Allendorf, 1986). En
phylogéographie, un tel phénomène peut impliquer une perte significative d’information car
ces allèles peuvent représenter des données historiques essentielles.
6La migration représente le déplacement des individus ou de leurs gamètes dans
l’espace. Le terme migration est généralement utilisé lorsque l’on réfère au déplacement
physique des individus, mais peut également faire référence au flux génique, soit à l’échange
d’allèles entre différentes populations ou différents taxons. Chez les espèces animales, la
migration est associée au déplacement des individus alors que chez les plantes, puisque
l’individu devient sessile une fois établi, la migration s’opère lors de la dissémination des
graines. En outre chez les plantes, le flux de gènes entre deux populations peut intervenir via
la dissémination du pollen et cela, sur de plus ou moins grandes distances. En règle générale,
le flux génique augmente la diversité des populations car il résulte en un apport d’allèles
exogènes.
La migration est parfois la cause d’événements fondateurs caractérisés par une chute
de la diversité lors de la colonisation d’un nouveau territoire par une fraction de la population
migratrice. Dans un contexte de colonisation postglaciaire, ce genre d’événement est attendu.
Le résultat net sur la diversité est semblable à celui d’une chute rapide de la population
effective par une mortalité accrue (Allendorf, 1986). On appelle ces événements des goulots
d’étranglements (« bottleneck »).
1.2.2 Propriétés génétiques des refuges glaciaires
Les refuges sont généralement les zones où la diversité génétique régionale est la plus
grande (Comes & Kadereit, 1998; Taberlet et al., 1998). En effet, lorsque les refuges sont
compris dans l’aire de répartition actuelle de l’espèce, les populations bénéficient
généralement d’un grand laps de temps sans subir de fluctuation démographique pour se
différencier. Cependant, les populations dans les refuges peuvent être soumises à des effets
de dérive génétique dépendamment de leur taille efficace. Dans ce cas, on observera une
perte en diversité génétique et une possible différenciation génétique. Les populations des
refuges situés à l’extérieur de l’aire de répartition actuelle n’ont pas d’équivalent
contemporain. Leur signature génétique ne peut donc qu’être inférée à partir de celle des
populations actuelles. Lors d’une colonisation rapide, les territoires colonisés à partir des
refuges ont généralement une diversité génétique plus faible puisqu’un sous-ensemble
aléatoire et partiel des allèles est transmis lors de la colonisation (goulot d’étranglement;
7(Ibrahim et al., 1996; Provan & Bennett, 2008). Par opposition, la diversité génétique peut
être maintenue lorsque la colonisation est lente et progressive.
1.2.3 Marqueurs génétiques
Une variation d’un nucléotide est appelée polymorphisme simple de séquence ou SNP
(Single Nucleotide Polymorphism). Il se produit lorsque l’ADN polymérase remplace une
base par une autre lors de la réplication. Le SNP peut être non-synonyme s’il engendre une
modification dans la protéine traduite et altère donc potentiellement le phénotype, ou
synonyme s’il n’engendre pas de modification de la protéine et du phénotype.
Les microsatellites ou SSRs (Simple Sequence Repeats) sont des répétitions d’un à
quelques nucléotides. La réplication de l’ADN peut causer l’ajout ou le retrait d’une des
répétitions de la séquence et donc créer du polymorphisme que l’on dénome insertion-
délétion ou indel. Ces marqueurs sont utiles en génétique des populations car leur taux de
mutation est relativement élevé (Di Rienzo et al., 1994). Il est donc fréquent de retrouver
plusieurs allèles pour un même locus dans une population. Les SSRs se retrouvent le plus
souvent dans des régions non codantes, qui sont souvent flanquées par des séquences
conservées à l’intérieur d’une espèce et parfois même entre les espèces voisines, ce qui
facilite le développement d’amorces universelles de PCR (Polymerase Chain Reaction)
(Tautz, 1989). Certains microsatellites du génome chloroplastique (cpSSRs) sont reconnus
pour être polymorphes chez de nombreux conifères (Vendramin et al., 1996; Parducci et al.,
2001)
Dans une étude phylogéographique où l’on cherche à retracer l’histoire d’une espèce,
il est important que les marqueurs utilisés soient le moins possible soumis à la sélection
naturelle, c’est-à-dire que leurs fréquences alléliques soient les plus représentatives des
phénomènes démographiques passés (Avise, 2000).
1.2.4 Analyse des données génétiques
Différents indices estimant la diversité génétique sont utilisés en génétique des
populations. La richesse allélique (A) représente le nombre moyen d’allèles à un ou plusieurs
loci dans une population. Le pourcentage de loci polymorphes (P) correspond au pourcentage
8de loci qui possèdent plusieurs allèles dans une population. On peut également évaluer
l’hétérozygotie attendue (He), soit l’hétérozygotie que devrait avoir une population si cette
dernière se comportait comme une population idéale (à l'équilibre d'Hardy-Weinberg).
L’hétérozygotie est calculée en fonction des fréquences alléliques dans une population où la
sélection des allèles qui seront transmis d’une génération à l’autre est complètement aléatoire.
On peut aussi estimer différents indices de différenciation génétique entre les populations,
qui mesurent la proportion de la variabilité génétique qui est due à la fragmentation d’une
grande population en plusieurs sous-populations. On trouve parmi ces indices le FST (Wright,
1949), le GST (Nei, 1973), le RST (Slatkin, 1995) et le NST (Pons & Petit, 1996), qui sont des
indices d’apparentement standardisés compris entre 0 à 1. Une valeur de 1 indique que la
différenciation entre les populations est complète, tandis qu’une valeur de 0 signifie qu’il n’y
a aucune différence entre les populations. Le FST et le GST sont des indices d’apparentement
qui ne tiennent compte d’aucun modèle de mutation et considèrent que chaque allèle est
génétiquement équidistant des autres (Nei, 1973). Le NST tient compte de la distance
génétique entre les formes alléliques observées (Pons & Petit, 1996). Le RST est un indice
d’apparentement qui tient compte de la répartition des allèles au sein des populations, mais
également de la distance génétique entre les allèles, soit du nombre de pas mutationnels
nécessaires pour passer d’un allèle à un autre. Il est typiquement utilisé pour les marqueurs
moléculaires contenant des répétitions (microsatellites, minisatellites), car il prend en compte
un modèle de mutation « pas à pas » caractéristique de ces marqueurs (Slatkin, 1995).
1.3 Les génomes cytoplasmiques des conifères
1.3.1 Génome mitochondrial
Chez les conifères, la mitochondrie possède un génome d’une taille qui dépasse les 4
Mb (Nystedt et al., 2013). Le génome mitochondrial est ordinairement haploïde (Birky et al.,
1989), c’est-à-dire qu’il ne possède qu’une copie de chaque locus. Relativement à l’ADNcp,
les mutations nucléotidiques s’y accumulent à un taux très bas chez les plantes (Wolfe et al.,
1987; Laroche et al., 1997; Jaramillo-Correa & Bousquet, 2003). En contrepartie, le
réarrangement des gènes y est possible et parfois fréquent (Jaramillo-Correa & Bousquet,
2005; Petit & Vendramin, 2007). Les polymorphismes de l’ADN mitochondrial sont
9généralement considérés comme neutres lorsqu’ils sont localisés dans l’ADN non-codant.
Les fréquences alléliques dans les populations ne sont alors pas influencées par la sélection
naturelle (Avise et al., 1987). Le fait qu’ils soient transmis maternellement, et donc
disséminés uniquement par la graine sur de courtes distances à chaque génération, permet
d’observer avec une plus grande résolution les structures géographiques anciennes. Ainsi,
l’ADNmt est considéré comme le génome de choix pour les études phylogéographiques chez
les conifères. Puisqu’il est haploïde, l’effet des processus démographiques sur la diversité
génétique et les fréquences alléliques est plus important que sur le génome nucléaire qui
possède plus d’une copie de ses gènes. Les effets anciens d’isolement génétique et de dérive
génétique seront donc plus facilement détectables à l’aide de polymorphismes du génome
mitochondrial.
1.3.2 Génome chloroplastique
Le chloroplaste des plantes photosynthétiques possède un génome variant de 120 kb
à 217 kb (Downie & Palmer, 1992). Chez les conifères, sa taille est d’environ 120 kb (Nystedt
et al., 2013; Yi et al., 2013). Le taux de substitutions nucléotidiques y est plus élevé que chez
le génome mitochondrial (environ 3 fois plus élevé) (Wolfe et al., 1987). Transmis
paternellement, l’ADNcp est disséminé séquentiellement par le pollen et ensuite par la graine
et peut donc voyager sur de longues distances. La structure géographique observée des
différents allèles de l’ADNcp sera donc généralement plus uniforme que celle de l’ADNmt,
car le flux génique est plus important et les allèles peuvent rapidement être disséminés sur
de grandes distances (Ennos, 1994; Petit et al., 2005).
1.4 Espèce à l’étude : le mélèze laricin
1.4.1 Le genre Larix
La divergence évolutive du genre Larix date de l’époque du Miocène. Le genre est
donc relativement récent dans l’arbre phylogénique des conifères (Wang et al., 2000). Il
comprend 11 espèces, dont trois sont retrouvées en Amérique du Nord (Larix laricina [Du
Roi] K. Koch, Larix lyallii Parl. et Larix occidentalis Nutt.). Ces trois espèces forment un
groupe phylogénétiquement distinct des autres espèces du genre Larix (Gros-Louis et al.,
102005). Le mélèze subalpin (L. lyallii) et le mélèze occidental (L. occidentalis) ont des aires
de répartition restreintes au sud-ouest du Canada et au nord-ouest des États-Unis,
respectivement. Les aires de répartition de ces deux espèces ne chevauchent pas celle du
mélèze laricin (L. laricina, Figure 1.2). Des études phylogéographiques ont été réalisées sur
diverses espèces de mélèze asiatiques et européennes comme Larix sibirica Ledeb.,
(Semerikov et al., 2007; Araki et al., 2008), L. gmelinii Rupr. (Khatab et al., 2008;
Polezhaeva et al., 2010), ainsi que L. cajanderi, L. sukaczewii et L. kaempferi (Araki et al.,
2008; Khatab et al., 2008). À l’heure actuelle, aucune étude phylogéographique n’a été
réalisée sur les espèces de mélèze du continent nord-américain. Quelques marqueurs
permettant d’identifier les différentes espèces ont été développés chez Larix pour les trois
génomes (Nadeem et al., 2003; Gros-Louis et al., 2005). Cependant, peu de polymorphismes
du génome mitochondrial ont été trouvés, ce qui indique que même au niveau interspécifique,
Larix possède une faible diversité chez ce génome.
1.4.2 Larix laricina
L'aire de répartition transcontinentale du mélèze laricin (mélèze d'Amérique, L.
laricina) s'étend longitudinalement des Maritimes et Terre-Neuve/Labrador jusqu’au Yukon,
allant du sud de la Nouvelle-Angleterre aux Grands Lacs, en passant par le Québec, l’Ontario,
quelques états du centre-nord des États-Unis d’Amérique, les provinces des Prairies
(Manitoba, Saskatchewan et Alberta) et une partie des territoires du Nord-Ouest (Figure 1.2).
À cela s’ajoute la partie centrale de l’Alaska qui est disjointe du reste de l’aire de répartition
(Ritchie, 1987). Une répartition aussi étendue fait du mélèze laricin une espèce d’intérêt en
phylogéographie comparative puisque son aire de distribution géographique chevauche
plusieurs facteurs de vicariance potentiels. De plus, L. laricina couvre l’aire de nombreuses
autres espèces boréales, ce qui peut permettre d’identifier des facteurs communs de
vicariance à l’échelle transcontinentale (Bermingham & Moritz, 1998; Jaramillo-Correa et
al., 2009). Le mélèze laricin se retrouve dans plusieurs assemblages forestiers comme espèce
compagne, dans des communautés dominées par d’autres espèces (ex. : épinette noire,
épinette blanche, sapin baumier). Son principal compétiteur est l’épinette noire qui est plus
compétitive. Il possède la capacité de pousser sur plusieurs types de sol, mais est plus souvent
11retrouvé dans les tourbières (Burns & Honkala, 1990). Les populations de mélèze sont en
général petites et discontinues (Cheliak et al., 1988).
Figure 1.2. Carte représentant l’aire de répartition des trois espèces du genre Larix en
Amérique du Nord. L’aire de répartition est montrée en vert pour Larix laricina, en bleu pour
Larix lyallii et en rouge pour Larix occidentalis.
1.4.2.1 Éco-physiologie de l’espèce
Lorsque l’on monte en latitude à travers la forêt boréale fermée nord-américaine, on
observe une transition du couvert forestier allant d’une forêt mixte, composée d'un mélange
de conifères et de feuillus, vers des peuplements de plus en plus dominés par les conifères
généralement sempervirents (à l'exception du mélèze). La température basse du sol ayant des
effets limitants sur la décomposition, la minéralisation et l’absorption de l’eau et des
nutriments, cette caractéristique de sempervirence permet de conserver les ressources
investies dans le feuillage lors de la saison de croissance et ainsi de réduire le stress sur
l’individu (Chabot & Hicks, 1982; Gower & Richards, 1990). Les mélèzes se différencient
des autres espèces de conifères par le fait qu’ils perdent leurs aiguilles pendant la saison
froide où l’ensoleillement est plus court. Ils arrivent cependant à coloniser les mêmes
environnements que les espèces à aiguilles pérennes. Il a été démontré que les espèces à
aiguilles décidues produisent des aiguilles avec un taux de photosynthèse plus grand par unité
12Vous pouvez aussi lire

















































