FOCUS SUR LES VOIES ORALES - Actualités en Onco-Hémato S PERRIAT Pharmacien IUCT-O Réunion du 2/06/2015
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Actualités en Onco-Hémato
FOCUS SUR LES VOIES ORALES
S PERRIAT Pharmacien IUCT-O
Réunion du 2/06/2015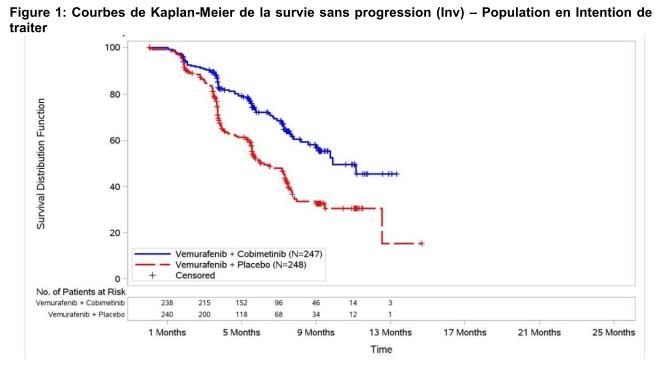
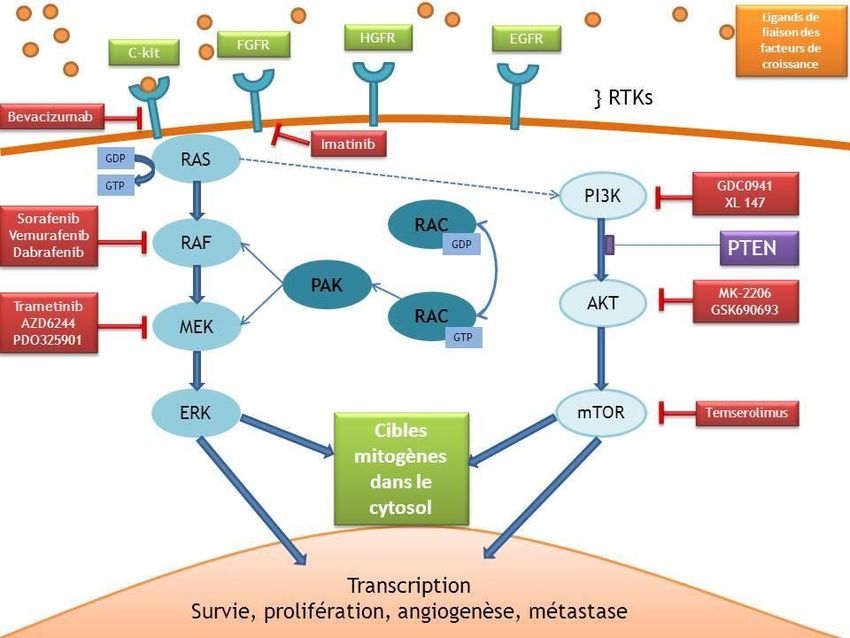
COBIMETINIB
• En association avec le vemurafenib (Zelboraf®) pour le traitement de patients
adultes atteints de mélanome non résécable ou métastatique, porteur d’une
mutation BRAF V600
• La dose recommandée : 60 mg une fois par jour pendant 21 jours sur 28
• EI Spécifiques : diminution FEV / augmentation CPK
• EI en asso Vemurafenib: Troubles oculaires -photosensibilité – éruptions cutanées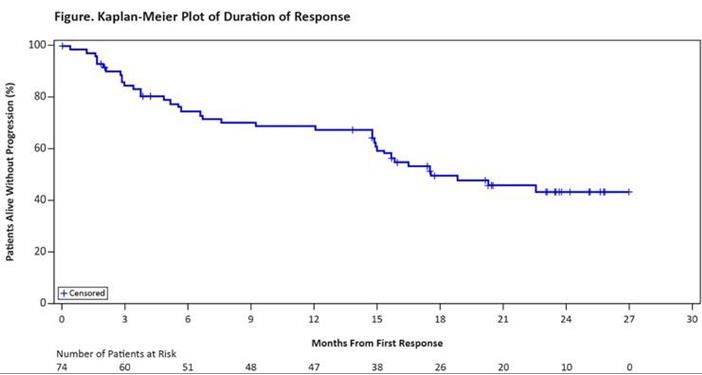
COBIMETINIB
Etude coBRIM - GO28141
• Phase III internationale , randomisée, en double aveugle, contrôlée versus placebo
• Objectif: d’évaluer la sécurité et l’efficacité du cobimetinib en association avec le
vemurafenib, par rapport au vemurafenib seul
• Critère principal : la survie sans progression (SSP) évaluée par l’investigateur
• Critères secondaires: survie globale (OS), taux de réponse objective et PFS évaluée
par un comité de revue indépendant (CRI)
– n= 495 patients après confirmation de la présence d’une mutation BRAF V600 à l’aide du
test de la mutation BRAF V600 cobas® 4800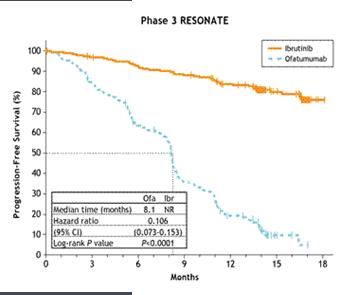
COBIMETINIB Etude coBRIM - GO28141
COBIMETINIB Etude coBRIM - GO28141
IBRUTINIB – Imbruvica ®
BTK signalisation via les récepteurs de surface des
cellules B
activation des voies enzymatiques nécessaires à
la circulation, au chimiotactisme et à l'adhésion des
cellules B.
Inhibition prolongée de l'activité
enzymatique
Inhibition de la signalisation du
récepteur antigénique des
cellules B (BCR) et du récepteur
des cytokines.
Ibrutinib inhibe efficacement la
prolifération et la survie des cellules
B malignesIBRUTINIB – Imbruvica ®
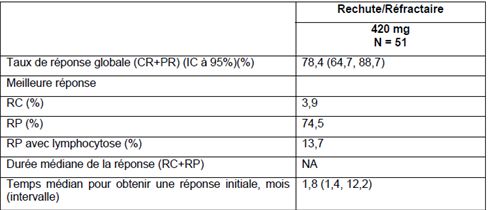
Etude PCYC-1104-CA: LCM en rechute ou réfractaire (AMM)
• Phase II multicentrique
• Objectif : Evaluation tolérance A 2 ans, OS = 47%
• Critère de jugement : OS selon investigateur SSP =31%
26% patients sous
traitement à 2 ans
81% patients EI G3
11% arrêt ttt suite EI
ASH 2014
ASH 2014 - M. Wang et al. Résumé ASH 4453IBRUTINIB – Imbruvica ®
LLC: 2 Etudes après au moins 1 ligne de traitement ou en 1ere ligne avec del 17p/TP53
• PCYC -1102-CA: Phase II en ouvert
• PCYC -1112-CA: RESONATE Phase III Multicentrique en ouvert n=391 patients
Tolérance:
76% des
patients
sous ttt à 16
mois
5% G3-4
Diarrhées
Nausées
Fatigue FAIBRUTINIB – Imbruvica ® 9
TP53 et inhibiteurs du BCR
• Phase II RESONATE™-17 p- traitement par ibrutinib
• n = 144 R/R patients avec del(17p) ORR = 82,6 %
SSP
Suivi médian : 13 mois
➜ À 12 mois, 88,3 % sont vivants dont 79 % en réponse
➜ Un Richter est observé chez 11 patients (7,6 %), 7 cas observés dans les 24 premières semaines de traitement
ASH 2014 - D’après O’Brien S at al., abstr. 327 ; Sharman J et al., abstr. 330, actualisésIBRUTINIB – Imbruvica ® 10
Perspectives
• Demande d’extension d‘AMM dans le cadre du traitement de patients adultes atteints de
macroglobulinémie de Waldenström (MW).
• Post allogreffe:
– L’efficacité de l’ibrutinib a été évaluée en post-allogreffe chez 5 patients en rechute clinique : 1 patient
avec une 17p- , traité pendant 37 mois, a une MRD négative et persistance de la réponse 8 mois après
l’arrêt de l’ibrutinib. Les autres patients sont toujours traités (3-17 mois) avec une excellente réponse
sur le syndrome tumoral
– L’utilisation de l’ibrutinib après allogreffe a été évaluée chez 16 patients dans les différents essais
thérapeutiques, 13 ayant reçu d’autres traitements pour la rechute post-allogreffe avant l’ibrutinib
Médiane de traitement : 18 mois
Réponse globale : 87,5 % avec 2 RC, 9 RP et 3 RC avec lymphocytose
Médiane de SSP à 2 ans : 77 % ; la seule cause d’arrêt du traitement a été une pneumopathie qui a
entraîné le décès chez 2 patients (à 0,9 et 2,3 mois)
• Essais cliniques: LBDGC,LF, MM
ASH 2014 - D’après Ryan E et al., abstr. 1186 ; Coutre S et al. abstr. 4697, actualisésIDELALISIB – Zydelig ®
PI3k:
hyperactivité dans
tumeurs malignes B
Prolifération,
survie,
migration et rétention
cel B dans tissus
lymphoïdeIDELALISIB – Zydelig ®
Etude Pivot 312-0116 LLC après au moins 1 ligne de ttt ou en 1ere ligne avec del 17p/TP53
• Phase III randomisée, double aveugle versus placebo
• Critère principal: survie sans progression (PFS)IDELALISIB – Zydelig ® 13
Idélalisib + rituximab : résultats actualisés chez les patients
en rechute ou réfractaires
Analyse de la SSP par sous-groupes
• Idélalisib + rituximab : pas d’impact 100 IDELA + R pas de del(17p)/TP53mut/del(11q)
sur la SSP des anomalies de TP53 ou 80
de la mutation du gène des immunoglobulines IDELA + R del(17p) ou TP53mut ou del(11q)
• SSP : 19,4 mois dans le bras RI versus 60
7,3 mois dans le bras rituximab + placebo 40 PBO + R pas de del(17p)/TP53mut/del(11q)
• À noter une augmentation des diarrhées 20
de grade 3-4 à 16 % versus 6 % dans la première PBO + R del(17p) ou TP53mut ou del(11q)
publication (Furman et al., N Eng J Med 0
0 2 4 6 8 10 12 14 16 18 Mois
2014;370(11):997-1007) • IDELA + R améliore la SSP, la RG et la survie également chez les
patients avec délétion ou mutation de TP53 (médiane de SSP :
16,6 mois versus 20,3 mois)
100
IDELA + R Médiane de SSP (mois) IGHV non mutées : idélalisib + R
80
IGVH mutées Non atteinte
60 IGHV mutées : idélalisib + R
IGVH non mutées 19,4
40
Pas de délétion 17p 20,3
20 IGHV mutées : placebo + R
Délétion 17p 16,6
IGHV non mutées : placebo + R Mois
0
0 2 4 6 8 10 12 14 16 18
ASH 2014 - D’après Sharman JP et al., abstr. 330, actualiséIDELALISIB – Zydelig ®
Etude - Pivot 101-09 LNH indolent (LF, LZM,MW) en monothérapie pour le traitement de
patients adultes atteints de lymphome folliculaire (LF) réfractaire à deux lignes de traitement
antérieures
• Phase III en ouvert
• Critère principal: ORR: proportion de sujets ayant obtenu une RC ou une
RP ou R mineure pour MW
• Critère secondaire: délai de réponse
– Interim results: ORR, 50.4% (95% CI, 41.3-59.5);
– SSP moyenne: 11.4 mois; durée moyenne de réponse: 11.9 mois.Ei (cutanés) peu décrits EC • Diarrhées retardées (à 9mois) grade III arrêt traitement • Toxicités cutanées type papulo-pustules • Prurit • FA • Hémorragies ++
OLAPARIB –Lymparza®
PARP = Poly ADP Ribose Polymérases
Famille d’enzymes
Nombreux rôles cellulaires, dont réparation dommages ADN :
Détecte cassures SB ADN Réparation
PARP
Protéines de
réparation
REPARATION DES
DOMMAGES A
L’ADNMécanisme d’action des Inhibiteurs de PARP (PARPi)
MAK, RTE
PARPi
PARP
PARPi
PARP
Mutation syst. réparation dont BRCA1/2
Chimio/Radio-sensibilisation Traitement K déficient en syst. réparation
Inhibiteurs de PARP + Anticancéreux Inhibiteurs de PARP en monothérapieOLAPARIB –Lymparza®
OLAPARIB :INHIBITEUR DE Poly ADP Ribose Polymerase (PARP)
• Monothérapie dans le traitement d'entretien des patientes adultes, atteintes d’un
cancer de l'ovaire récidivant (et des trompes de Fallope ou péritonéal primitif), ayant
une mutation BRCA, en réponse (complète ou partielle) à une chimiothérapie à base
de platine et qui, par ailleurs :
– sont sensibles à la chimiothérapie à base de platine (la sensibilité au platine est
déterminée par l’absence de progression de la maladie après une période de 6 mois (183
jours minimum) suivant la dernière cure de platine)
– ne peuvent pas être incluses dans un essai clinique en cours
– pour lesquelles il n’existe pas d’alternative thérapeutique appropriée
• La dose recommandée d’Olaparib est de 400 mg prise deux fois par jour (soit 16
gélules) à distance des repas (1h avant ou 2 h après)
• Les CYP3A4/5 sont les isoenzymes principalement responsables de la clairance
métabolique de l'olaparib.OLAPARIB –Lymparza® Etude ayant conduit à l’ATU • D0816C00002 (SOLO 2): Etude de phase III multicentrique, randomisée en double aveugle contrôlée avec placebo ayant pour but d’évaluer l’efficacité et la sécurité d’OLAPARIB comme traitement d’entretien chez des patientes adultes en rechute du cancer de l'ovaire avec mutation de BRCA sensible au platine et qui sont répondeuses (réponse complète ou réponse partielle) à une chimiothérapie à base de platine. • • D0816C00001 (SOLO 1): Etude de phase III multicentrique, randomisée en double aveugle contrôlée avec placebo ayant pour but d’évaluer l’efficacité et la sécurité d’OLAPARIB en traitement d’entretien chez des patientes adultes ayant un cancer de l'ovaire nouvellement diagnostiqué à un stade avancé (FIGO Stade III-IV) avec une mutation BRCA et étant en réponse complète ou réponse partielle à une chimiothérapie à base de platine en première ligne.
OLAPARIB –Lymparza®
• Merci de votre attention
Vous pouvez aussi lire

















































