Creative commons : Paternité - Pas d'Utilisation Commerciale - Pas de Modification 2.0 France (CC BY-NC-ND 2.0) ...
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous

http://portaildoc.univ-lyon1.fr
Creative commons : Paternité - Pas d’Utilisation Commerciale -
Pas de Modification 2.0 France (CC BY-NC-ND 2.0)
http://creativecommons.org/licenses/by-nc-nd/2.0/fr
BENSAHKOUN
(CC BY-NC-ND 2.0)
UNIVERSITE CLAUDE BERNARD - LYON 1
FACULTE DE PHARMACIE
INSTITUT DES SCIENCES PHARMACEUTIQUES ET BIOLOGIQUES
2019 THESE n°99
THESE
pour le DIPLOME D'ETAT DE DOCTEUR EN PHARMACIE
présentée et soutenue publiquement le 18 juillet 2019
par
M. BENSAHKOUN Julien
Né le 05 juillet 1989
à Lyon
*****
CONTRE-INDICATIONS MEDICAMENTEUSES DU THESAURUS DE L’ANSM RENCONTREES EN
OFFICINE
*****
JURY
M. TOD Michel, Professeur des Universités, Praticien Hospitalier
M. CATALA Olivier, Professeur des Universités et Personnel Associé à Temps Partiel
M. HALABI Jonathan, Docteur en Pharmacie
1
BENSAHKOUN
(CC BY-NC-ND 2.0)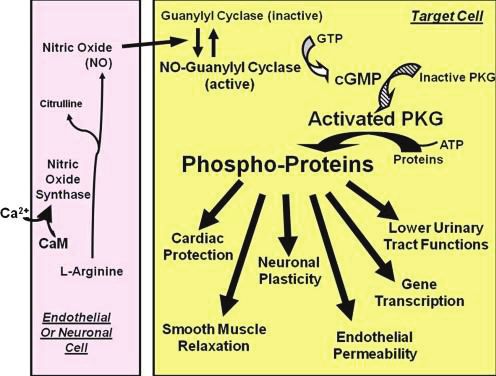
2
BENSAHKOUN
(CC BY-NC-ND 2.0)
UNIVERSITE CLAUDE BERNARD - LYON 1
FACULTE DE PHARMACIE
INSTITUT DES SCIENCES PHARMACEUTIQUES ET BIOLOGIQUES
2019 THESE n°99
THESE
pour le DIPLOME D'ETAT DE DOCTEUR EN PHARMACIE
présentée et soutenue publiquement le 18 juillet 2019
par
M. BENSAHKOUN Julien
Né le 05 juillet 1989
à Lyon
*****
CONTRE-INDICATIONS MEDICAMENTEUSES DU THESAURUS DE L’ANSM RENCONTREES EN
OFFICINE
*****
JURY
M. TOD Michel, Professeur des Universités, Praticien Hospitalier
M. CATALA Olivier, Professeur des Universités et Personnel Associé à Temps Partiel
M. HALABI Jonathan, Docteur en Pharmacie
3
BENSAHKOUN
(CC BY-NC-ND 2.0)
4
BENSAHKOUN
(CC BY-NC-ND 2.0)
5
BENSAHKOUN
(CC BY-NC-ND 2.0)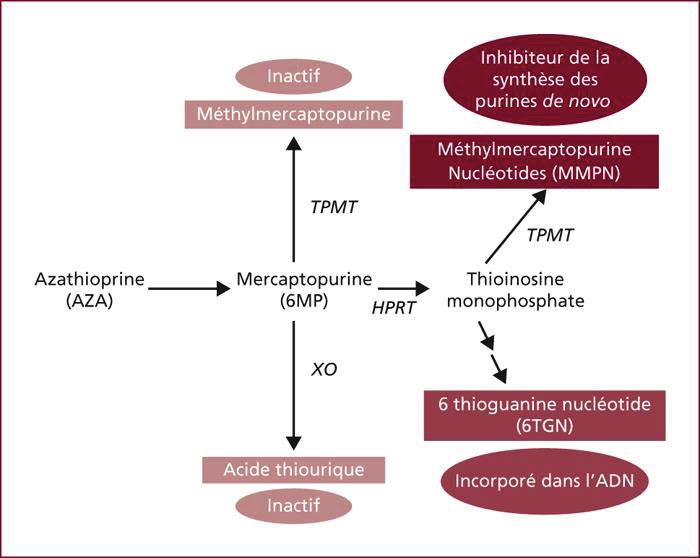
6
BENSAHKOUN
(CC BY-NC-ND 2.0)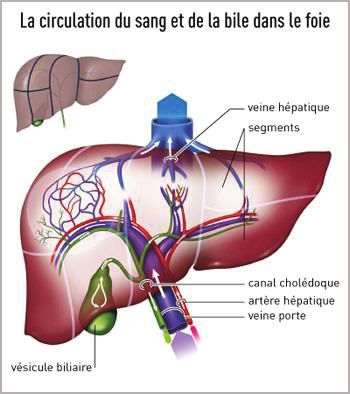
7
BENSAHKOUN
(CC BY-NC-ND 2.0)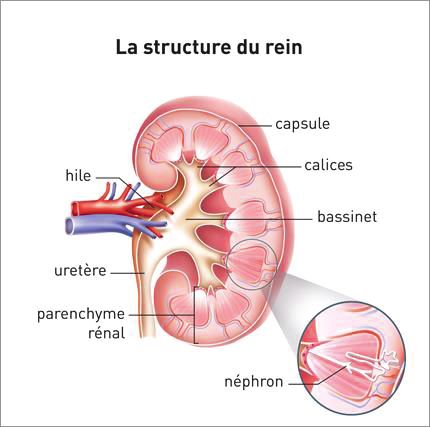
8
BENSAHKOUN
(CC BY-NC-ND 2.0)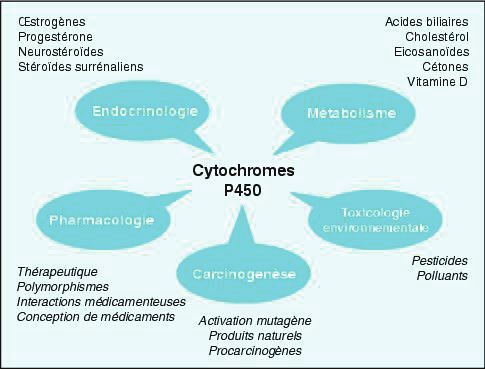
REMERCIEMENTS
Je tiens tout d’abord à exprimer mes sincères remerciements à M. Olivier Catala, pour
avoir dirigé cette thèse et m’avoir prodigué ses conseils avisés. Sa disponibilité et sa
gentillesse tout au long de ce travail méritent d’être distinguées.
Je remercie également M. Michel Tod pour sa réactivité et la présidence de cette thèse.
J’exprime par ailleurs toute ma reconnaissance à M. Jonathan Halabi, pour avoir porté
de l’intérêt à mon travail.
Quant à mes parents et à mes frères, je ne saurais les remercier pour l’amour et le
soutien qu’ils m’ont témoigné durant ces longues années d’étude.
J’ai une pensée particulière pour ma femme, qui a eu la patience de relire mon travail
et pour mon fils Aviel, qui a égayé mes pauses. Je suis ravi de pouvoir présenter cette thèse,
avant la venue au monde de ma fille, qui m’a donné la motivation de clôturer ce travail.
Je remercie l’ensemble de mes oncles et tantes, notamment Laurence et Daniel, pour
avoir été d’une aide précieuse tout au long de mon parcours universitaire.
J’exprime toute ma gratitude envers mes cousins et cousines, en particulier à ma
cousine Stéphanie, qui m’a ouvert la voie des études en pharmacie.
J’espère rendre fière par ce travail ma grand-mère adorée, que je porte dans mon cœur.
Je remercie aussi mes amis d’enfance, Jérémy et Yoni, pour leur soutien infaillible.
A mes amis de la faculté, Bob, David, Mimoun, Noelly et Mélanie, je leur souhaite de
réussir dans leur vie personnelle et professionnelle.
A tous ceux que je n’ai pas cité, merci de votre présence et soutien.
9
BENSAHKOUN
(CC BY-NC-ND 2.0)TABLE DES MATIERES
TABLE DES ABREVIATIONS............................................................................................ 12
TABLE DES FIGURES, TABLEAUX ET ANNEXES ...................................................... 14
INTRODUCTION .................................................................................................................. 15
RAPPELS SUR LE DEVENIR DU MEDICAMENT DANS L'ORGANISME............... 16
1. LES GRANDES FAMILLES D'INTERACTIONS MEDICAMENTEUSES....................... 19
1.1 Le cytochrome P450 ............................................................................................................... 19
1.1.1 Rappels sur le cytochrome P450.................................................................................................... 19
1.1.2 Rôles dans l'organisme du cytochrome P450................................................................................. 19
1.1.3 Interactions médicamenteuses et cytochrome P450....................................................................... 20
1.1.4 Les molécules concernées par le cytochrome P450 ....................................................................... 21
1.2 La glycoprotéine P.................................................................................................................. 21
1.2.1 Rappels sur la glycoprotéine P....................................................................................................... 21
1.2.2 Rôles dans l'organisme et mécanismes d'action de la glycoprotéine P .......................................... 22
1.2.3 Les interactions médicamenteuses et la glycoprotéine P ............................................................... 22
1.3 Les transporteurs organiques anioniques et cationiques......................................................... 23
1.3.1 Rappels sur les transporteurs organiques anioniques et cationiques .............................................. 23
1.3.2 Rôles dans l'organisme des transporteurs organiques anioniques et cationiques ........................... 23
1.3.3 Mécanismes d'action des transporteurs organiques anioniques et cationiques .............................. 24
1.3.4 Les molécules concernées par les transporteurs organiques anioniques et cationiques ................. 24
1.4 La tachycardie ventriculaire ................................................................................................... 25
1.4.1 Rappels sur la tachycardie ventriculaire ........................................................................................ 25
1.4.2 Mécanismes d'action et tachycardie ventriculaire .......................................................................... 27
1.4.3 Les molécules pouvant engendrer une tachycardie ventriculaire................................................... 28
2. LES CONTRE-INDICATIONS MEDICAMENTEUSES EN OFFICINE ........................... 29
2.1 Hépato-gastro-entérologie ...................................................................................................... 29
2.1.1 Les inhibiteurs de la pompe à protons ........................................................................................... 29
2.1.2 Les anti-émétiques ......................................................................................................................... 29
2.2 Oncologie................................................................................................................................ 30
2.3 Immunologie........................................................................................................................... 32
2.4 Hématologie............................................................................................................................ 35
2.4.1 Les anti-vitamines K ...................................................................................................................... 35
2.4.2 Les nouveaux anticoagulants oraux ............................................................................................... 35
2.5 Cardiologie ............................................................................................................................. 36
2.5.1 Les inhibiteurs de l'enzyme de conversion .................................................................................... 36
2.5.2 Les bétabloquants .......................................................................................................................... 37
2.5.3 Les antagonistes calciques ............................................................................................................. 38
2.5.4 Les anti-arythmiques...................................................................................................................... 39
2.5.5 Les autres classes thérapeutiques................................................................................................... 40
2.6 Endocrinologie........................................................................................................................ 41
2.6.1 Les antidiabétiques oraux .............................................................................................................. 41
2.6.2 Les hypolipémiants ........................................................................................................................ 42
2.6.3 L'inhibiteur de la parathormone ..................................................................................................... 43
2.7 Infectiologie............................................................................................................................ 43
2.7.1 Les antibiotiques ............................................................................................................................ 43
2.7.2 Les antiviraux ................................................................................................................................ 47
2.7.3 Les antifongiques ........................................................................................................................... 50
2.8 Neuropsychiatrie..................................................................................................................... 51
2.8.1 Les antidépresseurs ........................................................................................................................ 51
2.8.2 Les antimigraineux ........................................................................................................................ 58
2.8.3 Les anti-épileptiques ...................................................................................................................... 59
10
BENSAHKOUN
(CC BY-NC-ND 2.0)2.8.4 Les neuroleptiques ......................................................................................................................... 60
2.8.5 Les molécules utilisées en pharmacodépendance .......................................................................... 61
3. LES CONTRE-INDICATIONS AU SEIN DE POPULATIONS PARTICULIERES ......... 63
3.1 Chez l'insuffisant rénal ........................................................................................................... 63
3.1.1 Rappels .......................................................................................................................................... 63
3.1.2 Les rôles du rein............................................................................................................................. 64
3.1.3 L'insuffisance rénale ...................................................................................................................... 64
3.1.4 Les molécules favorisant l'insuffisance rénale fonctionnelle ......................................................... 66
3.1.5 Les molécules favorisant l'insuffisance rénale organique .............................................................. 66
3.2 Chez l'insuffisant hépatique.................................................................................................... 67
3.2.1 Rappels .......................................................................................................................................... 67
3.2.2 Les rôles du foie............................................................................................................................. 67
3.2.3 L'insuffisance hépatique ................................................................................................................ 68
3.2.4 Les mécanismes hépatotoxiques .................................................................................................... 69
3.2.5 Les molécules favorisant l'insuffisance hépatique ......................................................................... 69
CONCLUSION....................................................................................................................... 72
ANNEXES............................................................................................................................... 74
BIBLIOGRAPHIE ................................................................................................................. 77
RESUME................................................................................................................................. 90
11
BENSAHKOUN
(CC BY-NC-ND 2.0)TABLE DES ABREVIATIONS
UDP : Uridine diphosphate
CYP : Cytochrome P450
P-GP : Glycoprotéine P
MDR1 : Multi drug resistance 1
ATP : Adénosine triphosphate
SLC : Solute carrier family
OAT : Organic-anion-transporter
OATP : Organic-anion-transporting-polypeptide
OCT : Organic-cation-transporter
H+ : Hydrogène
Na+ : Sodium
K+ : Potassium
TV : Tachycardie ventriculaire
ECG : Electrocardiogramme
BAV : Blocs auriculo-ventriculaires
ANSM : Agence nationale de sécurité du médicament et des produits de santé
IPP : Inhibiteurs de la pompe à protons
AINS : Anti-inflammatoires non stéroïdiens
IL-1 : Interleukine 1
IL-6 : Interleukine 6
TNF- α : Tumor necrosis factor alpha
CRP : Protéine C réactive
6MP : 6-mercaptopurine
AVK : Antivitamine K
TXA2 : Thromboxane A2
COX1 : Cyclo-oxygénase 1
IEC : Inhibiteurs de l’enzyme de conversion
GMPc : Guanosine monophosphate cyclique
NO : Monoxyde d’azote
HMG-CoA réductase : Hydroxy-méthyl-glutaryl-coenzyme A réductase
12
BENSAHKOUN
(CC BY-NC-ND 2.0)ABCG2 : ATP-binding cassette super-family G member 2
LCR : Liquide céphalo-rachidien
Récepteurs 5HT1B/D : Récepteurs 5-hydroxytryptamine 1B et 1D faisant partie des
récepteurs sérotoninergiques
Peg-interferon α2 : Interféron pégylé alpha 2
ISRS : Inhibiteur sélectif de la recapture de la Sérotonine
IRSNa : Inhibiteur de la recapture de la Sérotonine et de la Noradrénaline
IMAO : Inhibiteur de la monoamine oxydase
COMT : Catéchol-O-méthyltransférase
GABA : Acide gamma-aminobutyrique
IRA : Insuffisance rénale aiguë
IRC : Insuffisance rénale chronique
DFG : Débit de filtration glomérulaire
MDRD : Modification of diet in renal disease
CKD-EPI : Chronic kidney disease-epidemiology collaboration
ALAT : Alanine aminotransférase
ASAT : Aspartate aminotransférase
GGT : Gamma-glutamyl transpeptidase
PAL : Phosphatases alcalines
Protéines NS3, NS4 et NS5 : Protéines non structurelles 3, 4 et 5
13
BENSAHKOUN
(CC BY-NC-ND 2.0)TABLE DES FIGURES, TABLEAUX ET ANNEXES
Figure 1 : Les différents rôles des cytochromes P450 dans l’organisme.………………...20
Figure 2 : Le mécanisme d’action de la P-GP……………………………………………..22
Figure 3 : Métabolisme de l’Azathioprine…………………………………………………33
Figure 4 : Le système rénine-angiotensine…………………………………………………37
Figure 5 : Métabolisme des dérivés nitrés…………………………………………………40
Figure 6 : Anatomie du rein…………………………………….…………………………..63
Figure 7 : Anatomie du foie………………………………………………………..………..67
Tableau I : Réactions de phase 2……………………………………………….…………..18
Tableau II : Contre-indications médicamenteuses avec le Millepertuis………………....57
Tableau III : Les différents stades de l’insuffisance rénale……………………………....65
Tableau IV : Mécanismes de toxicité hépatique et lésions hépatiques……………..…….69
Annexe 1 : Substrats des CYP 450 et de la P-GP………………………………………….74
Annexe 2 : Inhibiteurs des CYP 450 et de la P-GP………………………………………..75
Annexe 3 : Inducteurs des CYP 450 et de la P-GP………………………………………..76
14
BENSAHKOUN
(CC BY-NC-ND 2.0)INTRODUCTION
L’ANSM propose un outil d‘aide à la prescription et à la délivrance du médicament. Il
s’agit du thésaurus. Il regroupe toutes les interactions médicamenteuses qu’on peut rencontrer,
notamment en officine. Il est renouvelé tous les deux ans. Régulièrement des notes de mise à
jour sont publiées. On y retrouve la totalité des principes actifs par ordre alphabétique.
Chaque interaction y est répertoriée à deux emplacements différents.
Les interactions sont classées en quatre grades d’importance croissante : interaction
« à prendre en compte », précautions d’emploi, association déconseillée et contre-indication.
Pour les deux premiers grades, le pharmacien conseille au patient sur le bon usage du
médicament.
Pour les associations déconseillées, le pharmacien doit évaluer la balance
bénéfices/risques. Il s’assure de la bonne surveillance du traitement et vérifie si les examens
nécessaires à la surveillance sont bien prescrits et observés.
En cas de contre-indication, le pharmacien ne doit pas délivrer le médicament et se
doit de proposer une alternative aux médecins.
Pour une meilleure compréhension de ces contre-indications, nous avons explicité
dans une première partie les grandes familles d’interactions médicamenteuses puis détaillé les
contre-indications médicamenteuses rencontrées en officine dans une seconde partie. Enfin, la
troisième partie précise les contre-indications existant dans des populations particulières,
telles que chez les insuffisants rénaux et hépatiques.
L’objectif de ce travail est donc d’expliquer sur le plan pharmacologique les différentes
contre-indications recensées dans le thésaurus de l’ANSM, en mettant en exergue les
mécanismes pharmacocinétiques et pharmacodynamiques.
15
BENSAHKOUN
(CC BY-NC-ND 2.0)RAPPELS SUR LE DEVENIR DU MEDICAMENT DANS
L’ORGANISME
Dans ce chapitre, nous allons détailler les différentes étapes suivant l’administration
d’un médicament, à savoir son absorption, sa distribution, son métabolisme et son
élimination.
Absorption :
L’absorption d’un médicament est l’étape entre l’administration et le moment où il
rejoint la circulation sanguine. Puis le médicament va agir sur les organes cibles, tels que les
vaisseaux, le cœur, ou encore le cerveau.
Le plus souvent, le médicament est ingéré par voie orale. Dans ce cas, l’absorption se
passe au niveau de la muqueuse stomacale ou intestinale. Le passage des muqueuses peut se
faire par diffusion active grâce à des transporteurs ou par diffusion passive lorsque le
médicament est liposoluble.
Les perturbations du tube digestif influent sur le passage du médicament dans la
circulation générale. Une éventuelle diarrhée ou constipation peut réduire ou allonger le temps
de passage du médicament dans le tube digestif, ce qui peut faire varier son absorption. De
même, l’augmentation du pH gastrique par les inhibiteurs de la pompe à protons influe sur
l’ionisation du médicament et modifie son absorption.
Pour éviter ce problème, on peut administrer directement le principe actif dans la
circulation sanguine via la voie sous-cutanée, intramusculaire ou intraveineuse. Cette dernière
reste néanmoins une méthode invasive et parfois douloureuse. Mais elle a l’avantage d’assurer
une meilleure biodisponibilité et une action plus rapide (1).
Distribution :
La distribution du médicament se fait dans la circulation sanguine et dépend du
16
BENSAHKOUN
(CC BY-NC-ND 2.0)caractère plus ou moins lipophile de ce dernier.
Le médicament se fixe à des protéines plasmatiques, lui permettant d’arriver à son site
d’action. Parmi ces protéines plasmatiques, on peut citer notamment l’albumine, mais aussi
l’alpha-1-glycoprotéine acide qui fixe préférentiellement les médicaments basiques,
contrairement à l’albumine.
Plusieurs molécules peuvent être transportées par les mêmes protéines. Cela entraîne
un phénomène de compétition voire de saturation des protéines de fixation. Une des
substances peut alors ne pas être totalement liée à la protéine de fixation, ce qui peut
provoquer des effets indésirables.
Lors de la distribution, le médicament comporte une fraction liée aux protéines de
fixation et une fraction dite libre. C’est cette dernière qui va agir sur l’organisme suite à sa
transformation par différents complexes enzymatiques (2).
Métabolisme :
Le métabolisme est la transformation du médicament par l’organisme. Bien souvent,
cette étape implique des enzymes qui vont réagir avec le médicament et le métaboliser pour
favoriser son action. Le métabolisme comprend des réactions de phase 1 et de phase 2.
Dans les réactions de phase 1, on retrouve différents complexes enzymatiques. Le
principal est le cytochrome P450. Parmi les autres, on peut citer les époxydes hydrolases et les
flavines mono-oxygénases. Ces enzymes engendrent essentiellement des réactions
d’oxydoréduction, créant des métabolites polaires, souvent plus hydrophiles que la molécule
initiale. En règle générale, un groupe chimique est ajouté à la molécule mère, ce qui permet
l’action du métabolite. Mais il peut également y avoir des réactions d’hydrolyse.
Après les réactions de phase 1, on retrouve les réactions de phase 2 qui ajoutent une
fonction chimique ou modifient celles créées par la phase 1. Dans la phase 2 explicitée dans le
tableau I ci-après, s’effectuent des réactions de conjugaison, qui sont le plus souvent
réversibles, telles que la sulfonation, la glucuronidation, l’acétylation et la méthylation. Cela
engendre un complexe hydrosoluble, dont l’élimination rénale en sera facilitée (3).
17
BENSAHKOUN
(CC BY-NC-ND 2.0)Tableau I : Réactions de phase 2
Réactions de phase 2 Enzymes Localisations
Sulfonation Sulfotransférase Cytosol
Glucuronidation UDP-gluconyltransférase Réticulum endoplasmique
Acétylation Arylamine-N-acétyltransférase Cytosol et mitochondrie
Méthylation Méthyltransférase Réticulum endoplasmique, cytosol et sang
Elimination :
La principale voie d’élimination de l’organisme est la voie rénale, que nous
détaillerons plus particulièrement dans la troisième partie de cette thèse.
La deuxième voie la plus utilisée est la voie hépatique. Bien qu’elle soit le siège de
réactions de métabolisation, elle joue également un rôle non négligeable dans l’élimination
des médicaments. Après administration per os d’un médicament, celui-ci est absorbé par le
tube digestif puis dirigé vers le foie via la veine porte. Il est alors métabolisé et rejoint ensuite
soit la circulation générale par le biais de la veine cave inférieure, soit la bile. La bile peut à
son tour, soit être éliminée dans les selles, soit être réabsorbée en partie au niveau intestinal, à
l’origine du cycle entéro-hépatique (2).
18
BENSAHKOUN
(CC BY-NC-ND 2.0)1. LES GRANDES FAMILLES D’INTERACTIONS MEDICAMENTEUSES
1.1 Le cytochrome P450
1.1.1 Rappels sur le cytochrome P450
Le cytochrome P450 est une superfamille d’hémoprotéines située dans la membrane
du réticulum endoplasmique. On peut aussi le retrouver dans les hépatocytes, les cellules
intestinales, le rein, le poumon et d'autres tissus.
Son nom vient de sa capacité à absorber la lumière à une longueur d’onde de 450
nanomètres, lorsque le CYP 450 sous sa forme réduite Fe se lie au monoxyde de carbone.
Chez l'Homme, on retrouve environ 40 enzymes différentes, alors que plus de 200 ont
été répertoriées dans la nature. Une classification des isoenzymes composant le cytochrome
P450 a été établie. Par exemple lorsqu’on écrit CYP 3A4, CYP correspond au cytochrome
P450, 3 à la famille, A à la sous-famille et 4 à l'isoforme. Les différentes familles sont
classées selon le pourcentage d'analogie dans leur séquence d'acides aminés. Si le pourcentage
d’analogie est supérieur à 50 % entre deux molécules, elles appartiennent à une même famille.
Chez l’Homme, il existe 17 familles.
Nous allons nous intéresser plus particulièrement à quatre isoenzymes, responsables
d'environ 90% du métabolisme des médicaments. Il s'agit des cytochromes CYP 1A2, CYP
2C9, CYP 2D6 et CYP 3A4. Bien entendu on retrouve d'autre isoenzymes comme le CYP
2B6, le CYP 2C8, le CYP 2C19 et le CYP 3A5 dans certaines contre-indications.
Par ailleurs, certaines substances exogènes sont métabolisées par plusieurs
cytochromes et un même médicament peut être à la fois substrat et inhibiteur du même
cytochrome, ce qui complexifie les interactions. L’ensemble de ces interactions est
visualisable dans les annexes 1 à 3 (4) (5).
1.1.2 Rôles dans l’organisme du cytochrome P450
Le cytochrome P450 a différents rôles illustrés dans la figure 1.
Les mono-oxygénases du cytochrome P450 sont indispensables dans les
biotransformations de substances endogènes comme les stéroïdes, les acides gras, les
19
BENSAHKOUN
(CC BY-NC-ND 2.0)prostaglandines et les acides biliaires. Elles sont aussi impliquées dans le métabolisme
exogène des substances entrant dans l'organisme. Les familles 1, 2 et 3 des CYP 450 sont les
plus souvent concernées dans le métabolisme des médicaments.
Figure 1 : Les différents rôles des cytochromes P450 dans l’organisme (6)
1.1.3 Interactions médicamenteuses et cytochrome P450
Lorsqu’un médicament est métabolisé par une isoenzyme d'un cytochrome, on
l'appelle substrat de cette isoenzyme. Donc si l'on administre en parallèle un autre
médicament qui est inducteur ou inhibiteur de cette isoenzyme, cela provoque des
modifications de concentration plasmatique.
En cas d’association entre un inducteur et un substrat, le cytochrome va métaboliser
plus rapidement le médicament substrat et ainsi réduire sa concentration plasmatique, ce qui
diminue sa vitesse d'élimination. Inversement, lorsqu'on associe un inhibiteur à un substrat, on
a une augmentation de la concentration plasmatique du médicament substrat, car sa
métabolisation a été inhibée, et donc on observe une diminution de sa vitesse d'élimination.
20
BENSAHKOUN
(CC BY-NC-ND 2.0)Ces associations peuvent provoquer de graves conséquences, tels que des rejets de greffe ou
des surdosages (7).
1.1.4 Les molécules concernées par le cytochrome P450
Dans les annexes ci-jointes, nous avons listé les substrats, inhibiteurs et inducteurs des
cytochromes P450 et de la glycoprotéine P (8).
1.2 La glycoprotéine P
1.2.1 Rappels sur la glycoprotéine P
La P-GP produite suite à l'expression du gène MDR1, est un transporteur
membranaire, jouant un rôle important dans le devenir du médicament dans l'organisme. Elle
appartient à la superfamille des transporteurs ATP-binding cassette. Son poids moléculaire est
de 170 000 daltons et elle contient 1280 acides aminés. Comme d'autres protéines
transmembranaires, elle possède une partie intracellulaire hydrophile avec un site de fixation à
l’ATP et une partie intercellulaire hydrophobe composé de 6 hélices alpha (9).
On la retrouve essentiellement à la surface des cellules apicales du foie, des reins, de
l'intestin et du système immunitaire. Sa découverte s'est faite au sein des cellules tumorales.
Elle contribue en effet à la résistance aux traitements utilisés en chimiothérapie en provoquant
la sortie du médicament de la cellule. Sa localisation permet d'intervenir à des moments clés
du passage du médicament dans l'organisme, de son absorption à son élimination (10).
21
BENSAHKOUN
(CC BY-NC-ND 2.0)1.2.2 Rôles dans l'organisme et mécanismes d'action de la glycoprotéine P
Le rôle de la P-GP est de véhiculer les substrats à l’extérieur de la cellule. Pour établir
la liaison, le substrat se fixe au canal du coté cytosolique. Cela déclenche un changement de
conformation du transporteur afin de libérer le substrat.
La P-GP fonctionne contre un gradient de concentration afin de limiter l'absorption des
substances et notamment des médicaments dans le tractus digestif. Elle participe à
l'élimination des médicaments dans la bile et dans l'urine. Plus précisément, elle agit comme
une pompe en utilisant l'énergie de l'adénosine triphosphate, comme nous pouvons le
constater dans la figure 2. Son expression au sein de la barrière hémato-encéphalique
témoigne de son rôle de détoxification. Des travaux sur les souris ont en effet mis en évidence
que la P-GP bloque l’entrée des xénobiotiques dans le cerveau (11).
Figure 2 : Le mécanisme d’action de la P-GP* (12)
*Le triangle représente le médicament.
1.2.3 Les interactions médicamenteuses et la glycoprotéine P
Plusieurs stimuli peuvent provoquer une modulation de la P-GP (la chaleur, le stress,
les médicaments, les hormones...). Comme de nombreuses enzymes, la P-GP fonctionne grâce
22
BENSAHKOUN
(CC BY-NC-ND 2.0)à sa phosphorylation par une kinase. Les substances interférant avec cette kinase, perturbent
donc le bon fonctionnement de la P-GP.
La P-GP peut également subir des inductions et des inhibitions, comme le cytochrome
P450. Une induction du gène MDR1 entraîne l’augmentation de l'expression de la P-GP et
donc une diminution d'absorption des substrats par augmentation de leur transport vers
l'extérieur de la cellule. Inversement, l’inhibition de la P-GP augmente la concentration du
substrat à l'intérieur de la cellule, pouvant susciter un surdosage en substrat à l'intérieur de la
cellule (9).
1.3 Les transporteurs organiques anioniques et cationiques
1.3.1 Rappels sur les transporteurs organiques anioniques et cationiques
La superfamille SLC est composée de 52 familles qui regroupent environ 380
transporteurs. Leur poids moléculaire est d’environ 80 kilodaltons. Ces molécules sont
composées de 750 acides aminés, avec un domaine incluant 10 à 14 hélices alpha
transmembranaires. Leur localisation est ubiquitaire, mais on les retrouve plus
particulièrement dans les hépatocytes, les cellules intestinales, les reins, les poumons et le
cerveau.
Les transporteurs impliqués en pharmacologie sont peu nombreux. Il s’agit des
transporteurs OAT2, OATP1B1, OATP2B1, OATP1B3 et OCT1 (13).
1.3.2 Rôles dans l'organisme des transporteurs organiques anioniques et cationiques
Le rôle des ces transporteurs est opposé à celui de la P-GP. Ils ont pour objectif de
faire entrer les substrats à l’intérieur de la cellule. Les molécules anioniques sont transportées
par les transporteurs OATP lorsqu’elles ont une grande taille et par les transporteurs OAT,
lorsqu’elles sont plus petites. Concernant les cations, ils sont transportés par les transporteurs
OCT (14).
23
BENSAHKOUN
(CC BY-NC-ND 2.0)1.3.3 Mécanismes d'action des transporteurs organiques anioniques et cationiques
Le transport actif d’un médicament est couplé au transport d’ions tels que le H+ et le
Na+. Lorsque le médicament et ces ions vont dans des directions opposées, on parle de
transport antiport. A l’inverse, lorsqu’ils vont dans le même sens, c’est un transport symport.
Le transport antiport d’un substrat est couplé à un H+ ou à un autre ion. Pour
commencer, le substrat va rejoindre les acides aminés du canal externe des transporteurs afin
de s’y fixer. La fixation des ions provoque un changement de conformation du transporteur
qui va libérer le substrat à l’intérieur de la cellule.
Les transporteurs OAT et OATP sont fréquemment couplés à l’alpha-cétoglutarate.
Dans ce cas, la pompe Na+/K+-ATPase transfert à l’intérieur de la cellule deux molécules de
K+ contre trois molécules de Na+, créant un gradient de Na+. Le cotransporteur Na+ di-
carboxylate qui utilise ce gradient, va maintenir une concentration intracellulaire élevée de
l’alpha-cétoglutarate. Cela permet au transporteur du médicament anionique de faire entrer le
médicament à l’intérieur de la cellule en faisant sortir en contrepartie l’alpha-cétoglutarate.
Concernant le transport symport, il est habituellement couplé au Na+. Il utilise
l’énergie de la pompe Na+/K+ ATPase pour favoriser le changement de conformation du
transporteur à l‘arrivée du Na+ (13) (14).
1.3.4 Les molécules concernées par les transporteurs organiques anioniques et cationiques
Les transporteurs OATP1B1 :
Benzylpénicilline, Rifampicine, Méthotrexate, SN-38, Répaglinide, Troglitazone
métabolite, Fexofénadine, Bosentan, Valsartan, Olmésartan, Caspofungine, Atorvastatine,
Fluvastatine, Lovastatine, Pravastatine, Rosuvastatine et Simvastatine.
Les transporteurs OATP1B3:
Rifampicine, Docétaxel, Méthotrexate, Paclitaxel, Fexofénadine, Bosentan,
Olmésartan, Valsartan, Digoxine, Fluvastatine, Pravastatine et Rosuvastatine.
24
BENSAHKOUN
(CC BY-NC-ND 2.0)Les transporteurs OATP2B1:
Benzylpénicilline, Fexofénadine, Bosentan, Montélukast, Atorvastatine, Fluvastatine
et Rosuvastatine.
Les transporteurs OAT2:
Méthotrexate et Acide valproïque.
Les transporteurs OCT1:
Quinidine, Cisplatine, Imatinib, Oxaliplatine, Metformine, Cimétidine, Famotidine,
Ranitidine, Aciclovir, Ganciclovir, Lamivudine et Zalcitabine.
1.4 La tachycardie ventriculaire
1.4.1 Rappels sur la tachycardie ventriculaire
Contexte :
La TV est une augmentation régulière du rythme cardiaque à plus de 100 battements
par minute avec des complexes QRS larges (supérieurs à 120 millisecondes).
Son origine est ventriculaire, c'est-à-dire au-delà de la bifurcation hissienne.
La TV peut être monomorphe ou polymorphe, comme la torsade de pointes, son aspect
variant sur l'ECG.
Les étiologies de la TV sont les cardiopathies (ischémiques, valvulaires,
inflammatoires, congénitales), les cardiomyopathies, les troubles ioniques (dyskaliémie,
acidose), la iatrogénie (anti-arythmiques, antidépresseurs tricycliques) ou les excitants (café,
nicotine, alcool, cocaïne, amphétamines).
La TV engage le pronostic vital car elle peut se transformer en fibrillation ventriculaire
25
BENSAHKOUN
(CC BY-NC-ND 2.0)et en arrêt circulatoire. Son pronostic dépend de l'éventuelle cardiopathie sous-jacente (15).
Signes cliniques :
La TV peut engendrer des douleurs thoraciques, une dyspnée, des palpitations, une
tachycardie, une lipothymie ou une syncope (16).
Signes électrocardiographiques :
La TV est définie par plus de 3 extrasystoles ventriculaires successives (ESV). Une
ESV est caractérisé par un complexe QRS large et prématuré, non précédé d'une onde P.
La TV est dite soutenue si elle dure plus de 30 secondes.
Sur l'ECG, on peut retrouver une dissociation auriculo-ventriculaire (ondes P plus
lentes, dissociées des QRS), des complexes de capture (dépolarisation prématurée d'origine
supra-ventriculaire donnant un QRS fin) et de fusion (dépolarisation ventriculaire ectopique
donnant un QRS de largeur intermédiaire et de morphologie différente des autres), une
concordance positive ou négative (QRS entièrement positif (R) ou négatif (QS) de V1 à V6)
et une déviation axiale extrême en aVR.
Une torsade de pointes est un ensemble de complexes QRS d'amplitude et de polarité
variable, donnant un aspect en « accordéon » (17).
Torsades de pointes :
La torsade de pointes s'observe en cas d'allongement de l'intervalle QT et de
bradycardie lors des échappements à QRS larges des blocs atrioventriculaires bas situés, des
hypokaliémies ou hypocalcémies, de la prise de médicaments allongeant l'intervalle QT ou du
syndrome du QT long congénital (causes génétiques).
Elle peut s'arrêter spontanément ou dégénérer en fibrillation ventriculaire.
A savoir que les BAV complets peuvent engendrer des torsades de pointes (18) (19).
26
BENSAHKOUN
(CC BY-NC-ND 2.0)1.4.2 Mécanismes d'action et tachycardie ventriculaire
Il est difficile de lier un mécanisme d'action d'une molécule avec un trouble du rythme
spécifique. Les troubles du rythme sont en effet multifactoriels et certains anti-arythmiques
utilisent différents mécanismes d'actions. L'effet secondaire qui ressort le plus souvent est la
perturbation électrolytique causée par plusieurs classes de médicaments (les antipsychotiques,
antibiotiques et anti-arythmiques...).
Par exemple, l'hypokaliémie iatrogénique peut être causée par l'augmentation des
pertes digestives et urinaires et par l'activation du transport du potassium extracellulaire vers
le compartiment intracellulaire (20).
L'hyperkaliémie médicamenteuse peut provenir d'une insuffisance rénale ou
surrénalienne. Elle peut également être due à une acidose métabolique ou à une lyse cellulaire
(5).
Des facteurs de risques peuvent potentialiser les effets indésirables des molécules dites
torsadogènes.
Les facteurs de risque endogènes sont les métaboliseurs lents, le sexe féminin (la
repolarisation étant plus lente chez la femme), l’âge avancé, l’insuffisance hépatique et/ou
rénale, l’hypomagnésémie, l’hypo- ou l’hyperkaliémie, la bradycardie pré-existante (moins de
55 battements par minute), les anomalies génétiques des canaux potassiques et/ou sodiques, la
stimulation majeure du système sympathique, l'hypothyroïdie et une éventuelle cardiopathie
sous-jacente.
Les facteurs de risque exogènes sont l’utilisation de fortes posologies d'antibiotiques et
d'antipsychotiques ou encore les associations médicamenteuses augmentant l'intervalle QT
(21).
1.4.3 Les molécules pouvant engendrer une tachycardie ventriculaire
Certaines classes de médicaments favorisent le risque de perturbations électriques.
L'utilisation simultanée de deux médicaments torsadogènes est contre-indiquée en règle
générale.
27
BENSAHKOUN
(CC BY-NC-ND 2.0)Selon le thésaurus de l’ANSM, les médicaments suivants peuvent provoquer des
torsades de pointes :
Amiodarone, Amisulpride, Arsenieux, Chloroquine, Chlorpromazine, Citalopram, Cocaïne,
Cyamemazine, Disopyramide, Domperidone, Dronedarone, Droperidol,
Erythromycine, Escitalopram, Flupentixol, Fluphenazine, Halofantrine, Haloperidol,
Hydroquinidine, Hydroxychloroquine, Hydroxyzine, Levomepromazine, Lumefantrine,
Mequitazine, Méthadone, Moxifloxacine, Pentamidine, Pimozide, Pipamperone, Pipéraquine,
Pipotiazine, Prucalopride, Quinidine, Sertraline, Sotalol, Spiramycine, Sulpiride, Tiapride,
Torémifène, Vandétanib,Vincamine et Zuclopenthixol.
Toutefois l’utilisation de certains médicaments avec d’autres médicaments
torsadogènes est seulement déconseillée, en raison de leur caractère incontournable. Il s’agit
de la Méthadone, de l'Hydroxychloroquine, des antiparasitaires (Chloroquine, Halofantrine,
Luméfantrine, Pentamidine) et des neuroleptiques.
28
BENSAHKOUN
(CC BY-NC-ND 2.0)2. LES CONTRE-INDICATIONS MEDICAMENTEUSES EN OFFICINE
2.1 Hépato-gastro-entérologie
2.1.1 Les inhibiteurs de la pompe à protons
La Rilpivirine a besoin d’un pH acide pour que son effet soit optimum, son absorption
dépendant de l’acidité gastrique. L’augmentation du pH de l’estomac par les IPP entraîne
donc une diminution d’absorption de la Rilpivirine, expliquant la contre-indication à utiliser
ces deux médicaments ensemble (22) (23).
2.1.2 Les anti-émétiques
Aprépitant et Pimozide :
Le Pimozide a la capacité d’allonger l’intervalle QT et est métabolisé par le CYP 3A4.
Si on lui associe un inhibiteur puissant du CYP 3A4 comme l’Aprépitant, la concentration
plasmatique du Pimozide augmente et majore alors le risque de torsades de pointe. Cette
interaction est donc d’ordre pharmacocinétique avec des conséquences sur la conduction
cardiaque, bien que l’Aprépitant n’ait pas la capacité d’augmenter l’intervalle QT (24).
Dompéridone et Fluconazole :
Ces deux médicaments peuvent augmenter l'intervalle QT. En les associant, le risque
potentiellement vital d’allongement de l’espace QT est donc majoré (25) (26).
Ondansetron et Apomorphine :
L'Apomorphine agit principalement sur les récepteurs dopaminergiques. L'un de ses
effets secondaires est l’hypotension artérielle par vasodilatation. De même, l'Ondansetron
29
BENSAHKOUN
(CC BY-NC-ND 2.0)peut également engendrer une hypotension artérielle (27). L’association de ces deux
médicaments est donc contre-indiquée, même si les mécanismes de leur interaction ne sont
pas encore totalement élucidés.
2.2 Oncologie
Méthotrexate associé au Probénécide, à l’Acicrétine ou au Triméthoprime :
Le Probénécide est utilisé dans le traitement de la crise de goutte. Il est contre-indiqué
avec le Méthotrexate car il bloque l'élimination rénale du Méthotrexate, en inhibant sa
sécrétion tubulaire. De plus, il agit en se liant aux mêmes protéines plasmatiques que le
Méthotrexate. Le Probénécide provoque donc une augmentation de la concentration
plasmatique du Méthotrexate et favorise ainsi sa toxicité hématologique. Si l'interaction n'est
pas prise en charge à temps, on risque une pancytopénie (28).
On retrouve le même mécanisme d'interaction entre l’Acicrétine ou le Triméthoprime
et le Méthotrexate (29).
Méthotrexate et AINS :
Les AINS réduisent la sécrétion tubulaire du Méthotrexate, majorant sa toxicité
hématologique. Par ailleurs, les AINS peuvent interagir sur la liaison entre le Méthotrexate et
l’albumine et augmenter ainsi la fraction libre du Méthotrexate et donc sa toxicité (30).
L’association des AINS au Méthotrexate utilisé à forte dose (plus de 20 milligrammes par
semaine) est donc contre-indiquée.
Mifarmutide et AINS :
A dose élevée, les AINS inhibent le facteur d’activation des macrophages par
inhibition de la cyclo-oxygénase. Or l’effet antitumoral du Mifarmutide, utilisé dans le
traitement de l’ostéosarcome, est médié par l’activation des macrophages (31). Le
30
BENSAHKOUN
(CC BY-NC-ND 2.0)Mifarmutide agit en effet sur des cytokines pro-inflammatoires telles que l’IL-1, l’IL-6, le
TNF-α et la CRP. Les AINS à forte dose, diminuent donc l’effet anti-tumoral du Mifarmutide,
ainsi que la production d’ IL-1 et de TNF-α dans les monocytes (32).
Mifamurtide et immunosuppresseurs (Tacrolimus et Ciclosporine) :
Le Mifamurtide est également un immunostimulant. Son mécanisme d'action passe par
l'activation des macrophages, car ce médicament les rend tumoricides.
On en déduit qu'il ne faut pas associer des immunodépresseurs au Mifamurtide, car ils
vont gêner l'action immunostimulante recherchée (33).
Bléomycine et Brentuximab :
La Bléomycine, utilisée lors de chimiothérapies, provoque certains effets secondaires
en fonction de sa concentration, notamment toutes sortes de manifestions pulmonaires
(inflammation pulmonaire, pneumopathies, fibrose pulmonaire…). Ceci est dû à la faible
présence de la Bléomycine hydrolase dans le tissu pulmonaire, étant jusqu’à quinze fois
moins concentrée au niveau pulmonaire que dans d’autres tissus. La Bléomycine hydrolase est
une aminopeptidase qui désactive chimiquement la Bléomycine (34).
Lors de l’utilisation concomitante de Brentuximab et de Bléomycine les répercussions
pulmonaires se majorent. Les patients signalaient en effet fréquemment une toux et une
dyspnée et présentaient des infiltrats pulmonaires inflammatoires. On en conclue qu’il ne faut
pas associer la Bléomycine avec le Brentuximab (35).
Cytotoxique ou corticoïde associés à un vaccin vivant atténué :
Les cytotoxiques utilisés lors des chimiothérapies détruisent les cellules cancéreuses
mais également les globules blancs, ce qui favorise la survenue d’infections. De même, un
corticoïde majore le risque infectieux.
31
BENSAHKOUN
(CC BY-NC-ND 2.0)L’association d’un vaccin vivant atténué à un cytotoxique ou à un corticoïde est
donc contre-indiquée, car l’administration du vaccin pourrait avoir l’effet inverse à celui
recherché, c’est-à-dire provoquer une infection en cas de système immunitaire affaibli par le
cytotoxique ou le corticoïde (36).
Mitotane et Spironolactone :
La Spironolactone est un diurétique épargneur potassique et un antagoniste des
récepteurs à l'aldostérone. De plus, elle inhibe la production surrénalienne et ovarienne
d'androgènes (37).
Le Mitotane est utilisé pour traiter un carcinome surrénalien. Son métabolisme et son
mode d'action ne sont pas totalement connus. Cependant, on sait qu'il agit sur les zones
réticulées et fasciculées des glandes surrénales en bloquant la production d'androgènes et de
glucocorticoïdes (38).
Selon le thésaurus de l'ANSM, il y a un risque de blocage de l'action du Mitotane lors
de l’utilisation de la Spironolactone. Le mécanisme précis de cette interaction reste encore
inconnu, mais ces deux molécules ont des sites d'actions communs au niveau des glandes
surrénales.
2.3 Immunologie
Dans cette sous-partie, nous allons développer les contre-indications à associer
certains médicaments à certains immunosuppresseurs.
Mercaptopurine ou Azathioprine associés à un inhibiteur de la Xanthine-oxydase :
L'Azathioprine et la Mercaptopurine sont métabolisées par le foie, par trois voies
différentes, comme nous pouvons le voir dans la figure 3. L'une de ces trois voies utilise la
Xanthine-oxydase qui transforme la 6MP en acide thiourique, qui est un composé inactif. Si la
32
BENSAHKOUN
(CC BY-NC-ND 2.0)Xanthine-oxydase est inactive, il va y avoir une quantité plus importante de 6MP, qui lui est
un composé actif. Les inhibiteurs de la Xanthine-oxydase, tels que l’Allopurinol ou le
Febuxostat, ne peuvent donc pas être associés à la Mercaptopurine ou l’Azathioprine (39).
Figure 3 : Métabolisme de l’Azathioprine (40)
Anti-TNF-α et vaccin vivant atténué :
Les vaccins vivants atténués sont constitués d’agents infectieux atténués (virus ou
bactéries). Cette atténuation désirée peut parfois être incomplète voire inefficace.
En conséquence, si l’on associe un vaccin vivant atténué à un traitement
immunosuppresseur, comme les anti-TNF-α, on risque de provoquer une infection (41).
Ciclosporine et Bosentan :
La Ciclosporine est à la fois un substrat et un inhibiteur du CYP 3A4. En parallèle, le
Bosentan est à la fois un substrat et un inducteur enzymatique du CYP 3A4. L’association de
33
BENSAHKOUN
(CC BY-NC-ND 2.0)Vous pouvez aussi lire

















































