EEG et maladies dégénératives en période néonatale - LILLE Journée du GNPCLE 31 mai 2018 M-D Lamblin Hôpital Jeanne de Flandre
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
EEG et maladies dégénératives
en période néonatale
Journée du GNPCLE
31 mai 2018
M-D Lamblin
Hôpital Jeanne de Flandre
LILLE
Nouveau Né Une centaine de maladies héréditaires du métabolisme (MHM) actuellement connues sont à révélation néonatale Le néonatologiste est donc souvent en première ligne pour évoquer la MHM, organiser les prélèvements à visée diagnostique et débuter un traitement La survie et la prévention des séquelles neurologiques dépendent de la précocité du diagnostic et de la prise en charge
Classification des MHM :
3 catégories
Maladies d’intoxication
– Symptômes par accumulation aiguë ou chronique
– d’une substance le plus souvent normale : Aa, AO
Maladies du métabolisme énergétique
– Anomalie de production, de stockage ou
d’utilisation de l’énergie
Déficit de synthèse ou de dégradation des
molécules complexes
– Lysosomes, peroxysomes
– Golgi, Re
– Neurotransmetteurs, Purines, pyrimidines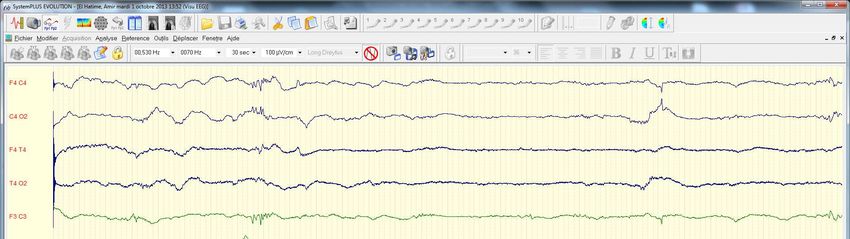
From Dr. Joe Clarke
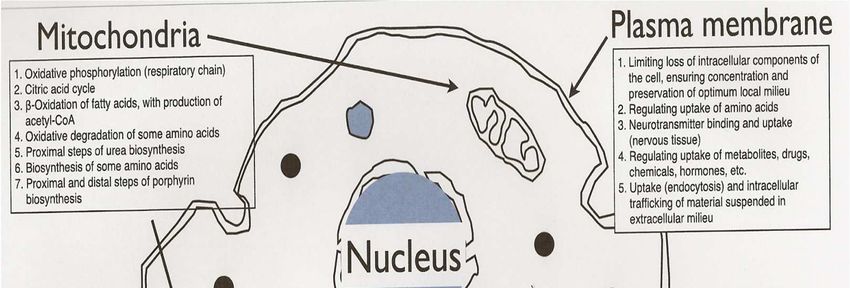
Fonctions du métabolisme
Synthèse et stockage d’Energie : Mitochondrie
Détoxification et recyclage : Lysosome
– Dégrade les molécules complexes pour les recycler
Logistique :
– Golgi, Réticulum endoplasmique
• Adressage : glycosylation des protéines
– Peroxysome : Organe de synthèse (acides biliaires, cholestérol,
plasmalogènes)
Communication : canaux, Récepteurs membranaires
– Intercellulaire
– Intracellulaire, entre organelles
– Interorganes : substances messagères métaboliques non hormonales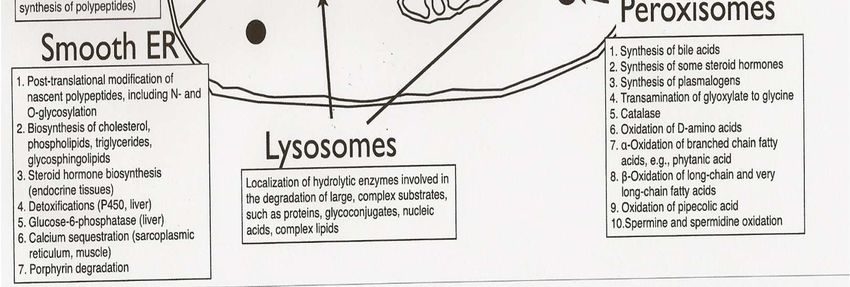
Classification des MHM : 3 catégories Maladies d’intoxication • d’une substance le plus souvent normale : Aa, AO • Symptômes par accumulation aiguë ou chronique Maladies du métabolisme énergétique • Anomalie de production, de stockage ou d’utilisation de l’énergie Déficit de synthèse ou de dégradation des molécules complexes • Lysosomes, peroxysomes • Golgi, Re • Neurotransmetteurs, Purines, pyrimidines
Maladies d’intoxication Accumulation aiguë ou chronique d’une substance normale : Protidique • Aminoacidopathies : Leucinose • Acidurie organique : Acidurie propionique, méthylmalonique • Déficit du cycle de l’urée Sucres complexes • Galactosémie
Leucinose Debut néonatal Coma et mouvements anormaux Bilan débrouillage « froid », acetone ++ CAAp +++ urgence Hemodiafiltration en urgence +++
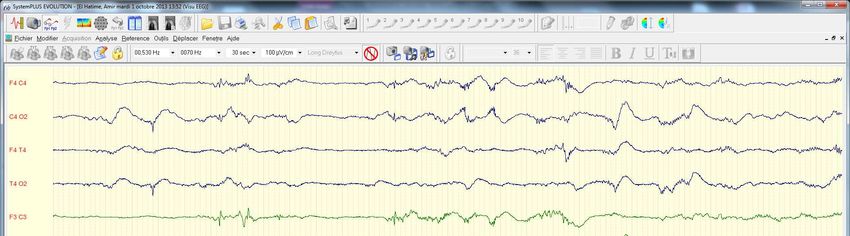
Amir El Ha.
er
Hypotonie 1 EEG ag 32 sem
J15
AC 33 sem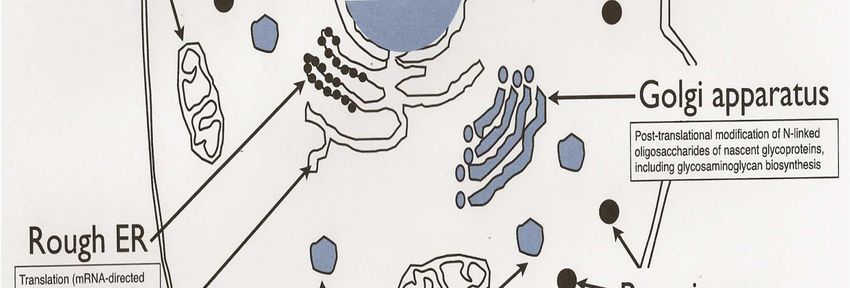
Ici diagnostic tardif car prema forte hypotonie d’où chromato AA
Maladies d’intoxication protidique Nouveau-né normal Intervalle libre Refus de boire, difficultés alimentaires Somnolence, léthargie Coma progressif Déshydratation
Maladies d’intoxication protidique
Coma
– Progressif
– S’agrave d’heure en heure
– Hypotonie axiale
– Hypertonie périphérique ++, opisthotonos
• Excitabilité
• Pédalage, boxe
• Myoclonies
• Trémulations de grande amplitude
• MI au-dessus du plan du lit
Tout coma avec une hypertonie ou un tonus normal doit
faire évoquer et rechercher une MHMMaladies d’intoxication protidique Comparer Ayoub et Lounis
Classification des MHM :
3 catégories
Maladies d’intoxication
• Symptômes par accumulation aiguë ou chronique
• d’une substance le plus souvent normale : Aa, AO
Maladies du métabolisme énergétique
• Anomalie de production, de stockage ou d’utilisation de l’énergie
Déficit de synthèse ou de dégradation des molécules
complexes
• Lysosomes, peroxysomes
• Golgi, Re
• Neurotransmetteurs, Purines, pyrimidinesMétabolisme énergétique :
Substrats énergétiques
Toutes les cellules nécessitent de l’ATP pour
assurer leurs fonctions
Substrats énergétiques :
• Glucose
‒ Tissus glycolytiques (glycolyse anaérobie)
• Acides gras (TG)
‒ Au repos : muscles et coeur
‒ Situations de demande énergétique accrue
• Effort prolongé, jeûne, fièvre, infections, hypoglycémieMaladies du métabolisme énergétique Anomalie de production, de stockage ou d’utilisation de l’énergie Atteintes pluri-viscérales mais surtout foie, muscle, coeur, cerveau hypoglycémie peut être le symptôme majeur : - Glycogénoses - Déficits de l’oxydation des AG hyperlactatémie peut être le symptôme majeur : Acidoses lactiques congénitales avec hypotonie majeure et polypnée – Déficit en pyruvate carboxylase (PC) – Déficit en pyruvate déshydrogénase (PDH) – Cytopathies mitochondriales
Cytopathie mitochondriale
glucose ADP
OH-but Ac Ac RCO2 Pi
Glycolyse : ATP ←
ATP
NAD+ NADH
OH-but/Ac Ac
LDH TCA
lactate pyruvate pyruvate
mitochondrie
NAD+ NADH
cytosolHypotonie néonatale et acidose
lactique
(PDH)
(PC)
Cytopathie mitochondriale1ere catégorie dans les
carences énergétiques
• Défaut d’oxydation des AG
• Cf Maya : hypoglycémie sans cétose
• Enz déficitaire : la translocase
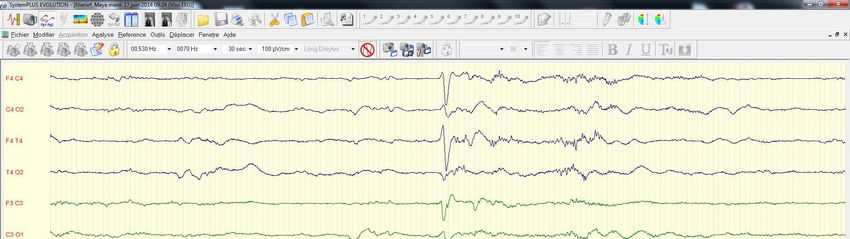
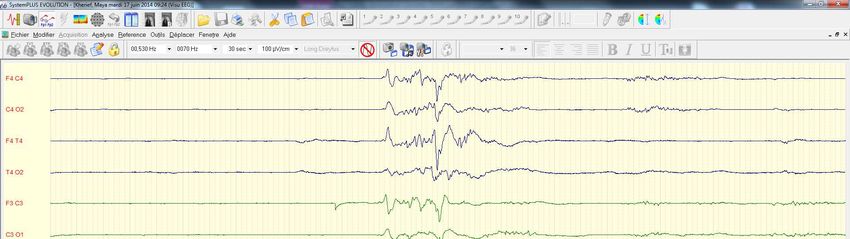
• Atteinte neuro tres sévèreKH.Maya dn 15 06 2014 • J2 • Hypoglycémie sévère, hypotonie, regard fixe • Cr convulsive • Troubles Béta oxydation
Vous pouvez aussi lire

















































