UE4 : Sciences thérapeutiques - Pr.Savary Pour toutes vos questions
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Faculté de santé d’Angers - département PluriPASS
Année universitaire 2020-2021
UE4 : Sciences thérapeutiques
Pr.Savary
Pour toutes vos questions : ue4@asso2atp.fr
Ledit polycopié a été entièrement réalisé par l’Association Angevine du Tutorat PASS (2ATP) avec
l’accord des enseignants référents. Ni les professeurs, ni la faculté ne peuvent être tenus responsables
de la validité des informations qu’il contient, même en cas de relecture par ces derniers.
Seuls les enseignements dispensés par les enseignants feront foi pour les examens.
Toute reproduction est interdite sans l’autorisation préalable de l’enseignant ou de la 2ATP.
Association Angevine du Tutorat PASS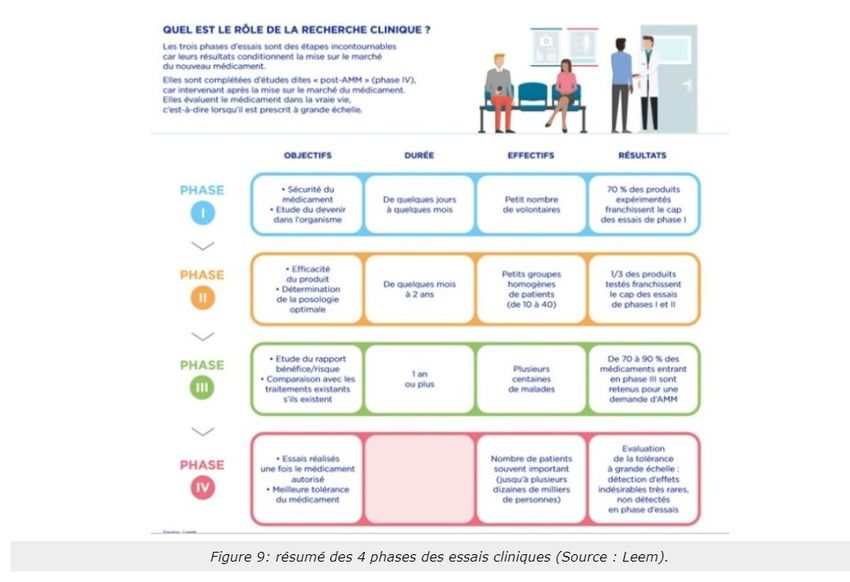
Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
SOMMAIRE
SCIENCES THERAPEUTIQUES ........................................................................... 2
I. Cours 1 : Toxicologie Pharmacovigilance ............................................................................ 2
Récapitulatif .............................................................................................. 20
Entrainements ........................................................................................... 21
Correction.................................................................................................. 22
Notes ........................................................................................................ 23
1
Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Sciences thérapeutiques
CHAPITRE N°10 : Prévention, sécurité et prise en charge du patient
I. Cours 1 : Toxicologie Pharmacovigilance
A) Généralités
1) Introduction
Dans cette partie, nous aborderons quelques définitions de base, la notion d'effet-dose, de bénéfice/
risque et de marge thérapeutique.
La prise en charge de la famille Tartempion nécessite, en autre, la prescription et la délivrance de médicaments.
Vous avez abordé dans les cours précédents, l'origine des principes actifs, les sites d'action possibles de ces
molécules à travers la pharmacologie, les voies d'administration ainsi que les formes galéniques disponibles
permettant une pharmacocinétique adéquate pour une meilleure efficacité possible.
Malgré un effet thérapeutique recherché, ces traitements peuvent engendrer des effets indésirables appelés
aussi effets secondaires, ou encore iatrogènes surtout en cas de surdosage.
2) Histoire
Paracelse disait : « tout est poison, rien n'est poison, c'est la dose qui fait le poison ». Il est considéré
comme le père de la toxicologie en formalisant la notion d'effet-dose (Figure 1). Pour une certaine quantité de
principe actif, les effets peuvent être bénéfiques induisant, ainsi, un effet thérapeutique alors que lorsque la dose
augmente, les effets indésirables peuvent apparaître. Ces effets peuvent être de simples nausées mais peuvent
être fatals et conduire à la mort de l'individu. Cette toxicité sera fonction de la molécule, de la dose administrée
et du patient (âge, comorbidité, etc...).
La discipline qui étudie ces effets est la toxicologie.
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
2
Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
3) Définition de la toxicologie – xénobiotique
La toxicologie est la discipline qui étudie les toxiques ou poisons : leur origine, leurs propriétés
physiques, chimiques et biologiques, leurs biotransformations, leurs modalités et mécanismes d'action sur
les systèmes vivants, leur détection et leur quantification, les moyens de combattre leurs actions nocives
par la mise en œuvre de procédés thérapeutiques appropriés et de mesure de prévention. Cette définition
très large permet de se rendre compte des multiples facettes et métiers en lien avec cette discipline (Source
Gatox).
Voici un exemple pour comprendre tous les enjeux de la toxicologie (ces informations ne sont pas à retenir mais
illustrent la définition) :
Le paracétamol peut être considéré comme un toxique puisque si la posologie n'est pas respectée, il induit des
effets indésirables importants).
« Son origine : Il s'agit d'une synthèse chimique : C4H6O3 + C6H7NO → C8H9NO2 + CH3COOH. Propriété
physique/chimique : la masse molaire peut être citée : 151,16 g/mol ; le pKa et de 9,382, la
T° de fusion de 169 à 171 °C ....
Biotranformation : l'ADME est parfaitement connu (cf cours de pharmacocinétique). L'absorption du paracétamol par
voie orale est complète et rapide. Le paracétamol se distribue rapidement dans tous les tissus. Le métabolisme est
hépatique et l'élimination se fait essentiellement par voie rénale. Cette biotransformation sera modifiée en cas de
surdosage. C'est ce qu'on appelle la toxicocinétique.
Effet biologique : effet antidouleur/ antipyrétique.
Détection et quantification : Il s'agit d'une méthode par spectrophotométrie. Le paracétamol est hydrolysé à chaud
en milieu acide (HCl) en donnant du para aminophénol. Ce dernier réagit avec le phénol en milieu alcalin en donnant
un dérivé de l'indophénol coloré en bleu qui permet un dosage spectrophotométrique.
Moyens de combattre leurs actions nocives : l'antidote du paracétamol est la N-acétyl cystéine. Des inscriptions sur
les boites de paracétamol et une posologie bien définie permet de prévenir des effets toxiques (cf exemple à la fin de
ce cours). »
La toxicologie s'intéresse aux poisons. Il s'agira de toute sorte de molécules qu'elles soient d'origines
naturelles ou synthétiques. Les toxiques peuvent être des médicaments mais également provenir de plantes,
de microorganismes (toxine de bactérie, champignons), d'animaux (venins) ou de molécules d'origine
industrielle. De manière générale, et les médicaments n'échappent pas à la règle, toute substance étrangère
au corps est appelée xénobiotique (= molécule étrangère au corps).
Les professionnels de santé seront bien évidements centrés sur les effets toxiques des médicaments mais
peuvent aussi être confrontés aux effets d'autres xénobiotiques, comme ceux de l'alcool, du tabac, des
intoxications professionnelles (amiante, peinture...), ménagères (ingestion accidentelle) ou encore aux
maladies développées suite à une exposition environnementale (cancer et pesticides, asthme et pollution
atmosphérique...). Dans le cadre de cette UE « Sciences appliquées à la thérapeutique », nous nous
concentrerons seulement à l'étude de la toxicité des médicaments. Pour ces médicaments, la notion de
bénéfice/risque est essentielle.
3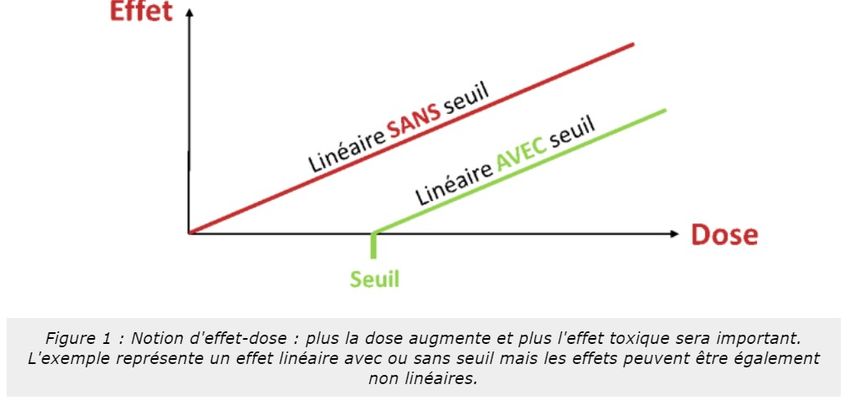
Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
4) Notion de bénéfice/risque
Chaque médicament est prescrit et délivré avec une recommandation de dose, appelée posologie.
Lorsque cette posologie n'est pas respectée et est revue à la baisse, l'effet thérapeutique souhaité ne sera pas
optimal. Dépassée, ce sont les effets indésirables ou toxiques qui peuvent apparaître. Vous avez vu cela avec M
Legeay en pharmacométrie. Parfois, à dose thérapeutique, les médicaments peuvent induire des effets
indésirables (appelés aussi effets iatrogènes). Ainsi nous parlons du rapport bénéfice/risque (= évaluation des
effets bénéfiques thérapeutiques en comparaison aux risques liés à la sécurité d’emploi d’un médicament. Il est
mesuré par une utilisation donnée ou estimés pour une population) qui sera évalué et acceptable ou non. Il s'agira
de comparer la dose efficace et la dose toxique et d'évaluer la marge ou index thérapeutique (cf ci-dessous figure
2).
Un peu de réflexion :
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
Dans ce graphique simplifié, sont représentées deux courbes en fonction de la dose (Figure 2). La courbe bleue
correspond à l'efficacité (effet thérapeutique) tandis que la courbe rouge représente les effets toxiques. La dose
efficace 50 est la dose nécessaire pour induire 50% de l'effet pharmacologique. La dose létale 50 est la dose
nécessaire pour induire 50 % de mortalité dans une population. Ici l'effet toxique est la mort, mais il aurait pu être
représenté par un effet indésirable comme la toxicité hépatique, ou encore par des douleurs gastriques ou des
vomissements etc...
4
Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
Dans ce graphique, lorsque l'efficacité est de 50 %, il n'y aura aucune mortalité. Si nous souhaitons obtenir 100 %
d'efficacité, malheureusement 50 % des personnes mourront. Dans ce cas, le médicament sera utilisé à la dose
efficace 50 s'assurant, ainsi, d'aucun effet toxique.
L'important est de connaître la marge ou index thérapeutique de chaque molécule (Figure 3). Il s'agira de la
différence entre dose efficace et dose toxique et permettra de connaître le rapport bénéfice/risque
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
Par ailleurs, les réponses à ces questions sont nécessaires pour déterminer l'acceptabilité de ce rapport :
• Existe-t-il un autre traitement plus efficace avec les mêmes effets indésirables ?
• Excite-t-il un traitement aussi efficace mais avec moins d'effets indésirables ?
• Les effets indésirables engendrés par rapport aux conséquences de la maladie sont –ils acceptables ?
5) Marge thérapeutique étroite
Pour certaines pathologies, il existe des traitements où les effets thérapeutiques sont obtenus à des doses
se rapprochant de la dose engendrant des effets iatrogènes ou toxiques. Ces médicaments, parfois seuls sur le
marché pour une pathologie donnée, sont appelés les médicaments à marge thérapeutique étroite (MTE :
s’emploie pour un médicament dont la dose thérapeutiques et la dose toxique sont proches et dont les
concentrations sanguines peuvent varier). Cela signifie que toute variation de sa concentration dans votre
organisme, même légère, peut éventuellement entraîner des effets indésirables, potentiellement graves. Ainsi,
la différence entre dose efficace et dose toxique est faible. Une attention particulière est indispensable lors de
leur délivrance.
5
Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Exemple : La ciclosprine est un immunosuppresseur utilisé dans la plupart des protocoles de transplantation (rein,
foie, cœur, poumon, pancréas, intestin grêle, moelle osseuse) afin d'améliorer la survie du greffon et de prévenir
la réaction du greffon contre l'hôte. C'est un médicament à marge thérapeutique étroite qui nécessitera une
surveillance par un dosage sanguin de cette molécule. En effet cette molécule est néphrotoxique (atteinte rénale)
et cette toxicité est directement liée aux concentrations sanguines obtenues (et non à la posologie). Celle-ci sera
différente d'une personne à une autre en fonction de la pharmacocinétique (cf cours M Legeay).
Le dosage sanguin permet de rééquilibrer la posologie pour rester dans la zone thérapeutique (Figure 3)
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
6) Pas si simple
Tout n'est pas si simple, car en dehors des effets induits par une molécule, il faudra prendre en compte
d'autres paramètres indépendants de la molécule : la pathologie indiquée (gravité, issus : gastroentérite/
grippe/cancer...), l'âge du patient (nourrissons/enfants/adultes/personnes âgées) et la comorbidité (autres
atteintes) et enfin les éventuelles interactions avec d'autres molécules médicamenteuses ou non, prises par
le patient.
Revenons sur ces trois éléments
1. Pathologie pour laquelle le traitement est indiqué : Le rapport bénéfice/risque prendra en compte la gravité
de la maladie. Un médicament aux nombreux effets indésirables ne sera pas accepté pour une pathologie bénigne
comme des maux de gorges. Au contraire, pour une pathologie sans issus comme certaines formes de cancer, les
effets secondaires seront plus « acceptés ».
« La grand-mère Tartenpion souffre d'une leucémie aigüe lymphoblastique. Pour cette patiente de plus de 60 ans chez
qui seule une chimiothérapie d'entretien peut être entreprise, la survie à 5 ans n'excède pas 15 à 20%. Néanmoins
sans traitement l'évolution est fatale. Les traitements mis en place ici sont la vincristine et la cytarabine. Ce sont deux
molécules cytotoxiques. L'objectif de ces molécules est de tuer les cellules sanguines qui sont tumorales.
6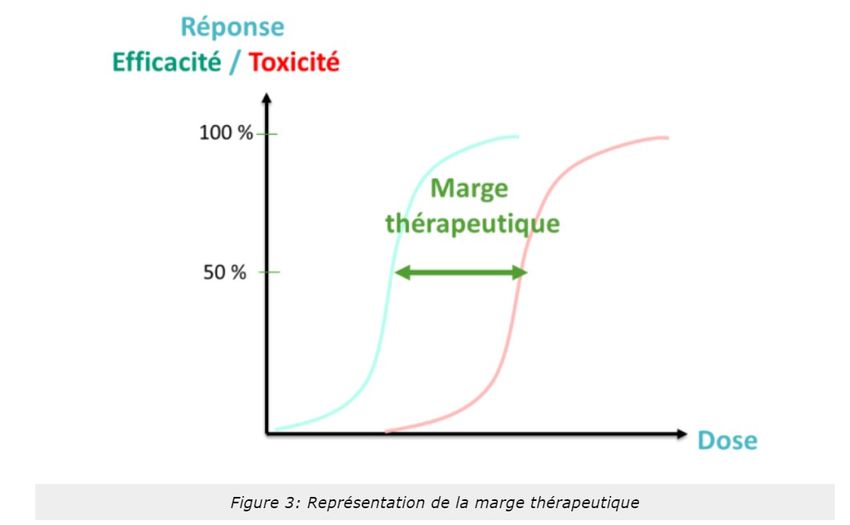
Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
Malheureusement, de par leur action, ces molécules vont également être cytotoxiques pour les autres cellules
sanguines non tumorales, engendrant ainsi des thrombopénies (manque de plaquette dans le sang engendrant des
hémorragies par exemple), et des cellules responsables de l'immunité. Cela peut contribuer au développement de
candidoses (développement de champignons pathogènes). Au regard de la gravité de la maladie, ces traitements
sont acceptés bien que les risques soient très élevés dans la mesure où le bénéfice l'est d'autant plus. Il est à noter que
des traitements peuvent aussi être ajoutés à la prescription de la patiente pour empêcher ces effets secondaires ou
au moins les traiter. C'est le cas avec la Caspofungine ici qui est utilisée pour traiter la candidose. Des conseils non
médicamenteux pourront être délivrés également par les professionnels de santé accompagnant ces patients
(pharmaciens, médecins généralistes, infirmiers) »
2. Comorbidité : les patients peuvent être atteints de plusieurs pathologies. Par exemple, un patient peut souffrir
d'une pathologie cardiaque et d'une insuffisance rénale. Un traitement peut être bénéfique pour un patient et
délétère si celui-ci est donné à un patient atteint d'une autre pathologie. Les insuffisances hépatiques et rénales
seront par exemple à prendre au sérieux car directement influentes sur la concentration sanguine en principe
actif. Des effets toxiques peuvent, ainsi, apparaitre même à dose thérapeutique.
3. Interaction médicamenteuse ou non. Certaines molécules (d'origine médicamenteuse, alimentaire ou
environnementale) peuvent influencer la pharmacocinétique lors des 4 phases (absorption, distribution,
métabolisme et élimination) mais aussi la pharmacodynamie (compétition par exemple) engendrant ainsi des
effets indésirables non prévus.
Exemple : Le jus de pamplemousse est connu pour inhiber un cytochrome P450 3A4 impliqué dans le métabolisme de
la simvastatine. Le père qui souffre d'hypertension artérielle prend ce médicament pour réduire sa tension artériel →
échec thérapeutique s'il boit du jus de pamplemousse tous les matins. (Voir la partie d'inhibition enzymatique du cours
de pharmacocinétique, Samuel Legeay)
Fort heureusement pour les patients, tous les médicaments disposent d'une certaine sécurité. En effet
que ce soit en amont de la mise sur le marché ou après grâce à la pharmaco/toxicovigilance, les effets
indésirables/toxiques de ces molécules sont évalués tout au long du cycle de vie du médicament ainsi que ce
rapport bénéfice/risque
B) L’évaluation de la sécurité du médicament lors de son cycle de vie
1) Introduction
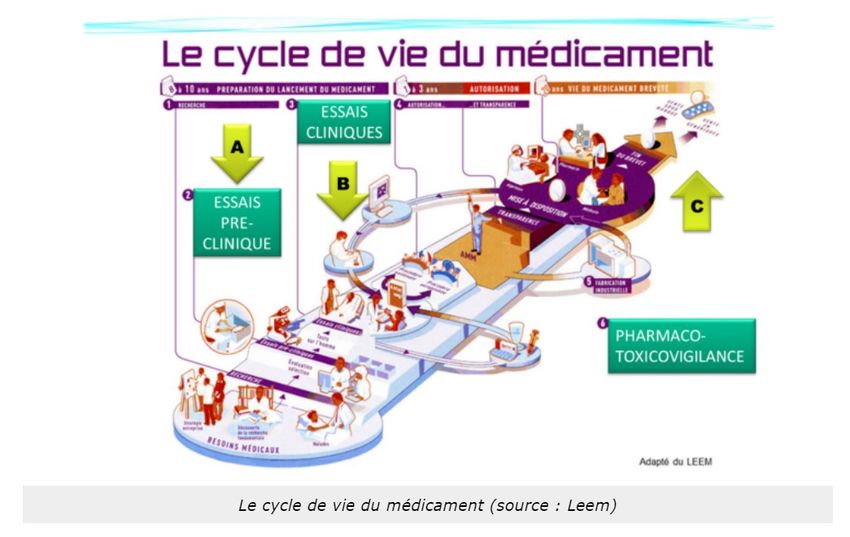
Voici le cycle de vie d'un médicament : Tout développement de médicaments provient d'un besoin vis-à-
vis des patients (diagnostic, vaccins, thérapie...).
Vous avez abordé avec Monsieur Papon et Madame Baglin, l'origine multiple des médicaments (naturelle ou
synthétique). Mais avant d'obtenir des médicaments qui permettent de diagnostiquer, de prévenir (vaccin) et de
traiter les pathologies de la famille Tartempion, le chemin est long...très long (Figure 5).
7Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
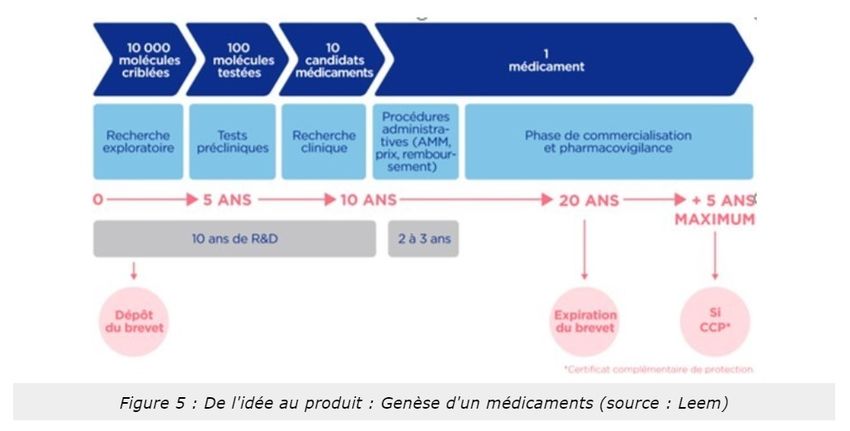
VIDEO EXPLICATIVE : Le développement d’un nouveau médicament est très réglementé. C’est aussi le résultat
d’un processus couteux, car il représente un investissement de prêt d’un milliard d’euros, et long, car il dure en
moyenne 10 ans.
Tout d’abord, il y a une recherche exploratoire, le laboratoire sélectionne toutes les molécules qui pourraient
guérir complètement les patients ou atténuer les effets d’une maladie ciblée. Une fois sélectionné le laboratoire
teste chacune des molécules sur des cellules et des animaux. Ce sont les tests pré-cliniques. Suite à ceci, une
molécule appelé candidat médicament est choisie pour être testé sur l’homme. Ce sont les essais cliniques en
trois phases (1-tolérence ; 2-efficacité ; 3-rapport bénéfice/risque). En parallèle, le laboratoire détermine le mode
d’administration du candidat médicament puis le produit. Si les essais sont satisfaisants, le laboratoire dépose un
dossier d’AMM (autorisation de mise sur le marché) qui décrit tous les résultats des essais. Ce dossier est transmis
à l’EMA (européen medecine agency) qui étudie le rapport bénéfices/ risques du médicament et en fonction du
résultat autorise ou non ça commercialisation. Cette étape dure environ 1 an. Une fois commercialisé, le
médicament est surveillé par le laboratoire et par les autorités sanitaires pour vérifier son efficacité et sa tolérance
en vie réelle. Le laboratoire peut ensuite le développer pour soigner d’autres maladies. C’est une extension
d’indication. Il peut aussi l’adapter pour les enfants.
Le respect de toutes ces étapes garantis qualité, sécurité, efficacité des médicaments mis sur le marché.
Alors, quand et comment les effets indésirables et la toxicité d'un médicament sont-ils déterminés ?
8Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
A. Lors des essais précliniques in vitro (sur cellules) ou in vivo (sur animaux),
B. Lors des essais cliniques sur des personnes saines puis un petit groupe de patients
C. Après son autorisation de mise sur le marché grâce à la pharmacovigilance sur une population de patients à
plus grande échelle.
Les deux premières phases (A et B) sont indispensables pour obtenir une autorisation de mise sur le marché
appelée couramment AMM, le troisième (C) pour le maintien de cette AMM.
2) Les études précliniques
GENERALITES
Les études précliniques permettent d'obtenir les premières connaissances sur le comportement d'un
candidat médicament issu des phases de la recherche expérimentale, indispensable avant les essais chez
l'Homme.
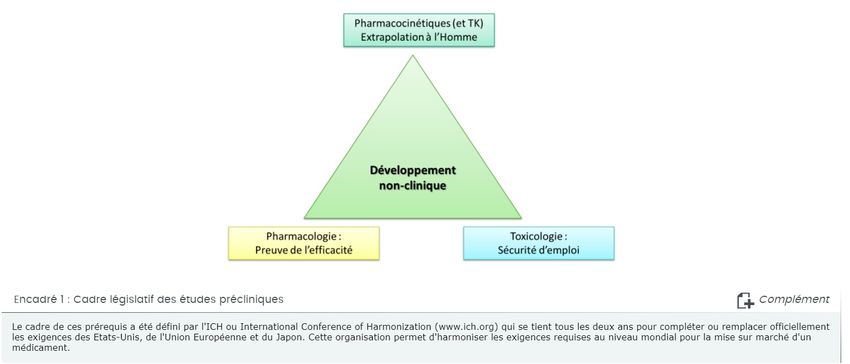
Au cours du développement préclinique, un grand nombre d'études est effectué afin de qualifier le candidat
médicament sur le plan de la pharmacologie, de la pharmacocinétique et de la toxicologie. Ces études sont
constitutives d'une partie du dossier de demande d'autorisation de mise sur le marché (AMM) du futur
médicament ; elles répondent à des normes internationales de qualité scientifique (Encadré 1) et sont
étroitement évaluées par les autorités de santé au moment de délivrer l'AMM.
9Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Le développement préclinique fait en particulier appel à l'expérimentation animale, qui est une étape
indispensable à la connaissance d'un futur médicament avant de l'administrer à l'Homme. En effet, il n'est pas
envisageable d'administrer un nouveau composé à l'Homme sain ou malade compte tenu des risques non connus
susceptibles d'apparaître. L'expérimentation animale est donc utilisée de manière rationnelle et, dans tous les
cas, selon des bonnes pratiques qui garantissent un traitement éthique de l'animal de laboratoire (Encadré 2).
Source : LEEM
Les études de pharmacologie (Figure 7) ont pour but de valider le mécanisme d‘action et de mesurer l'activité
du candidat médicament dans des modèles expérimentaux de la maladie, in vitro et in vivo. Les études in vivo
permettent d'évaluer l'activité pharmacologique, y compris de définir les mécanismes d'action, pour justifier
l'usage proposé du produit dans les études cliniques. Ce sont ainsi les mécanismes d'actions et l'efficacité qui sont
recherchés sur la base de ce que vous avez vu dans le cours de pharmacologie de Monsieur Legeay. Dans cette
partie, est étudiée également la pharmacologie de sécurité. « Apparenté à la toxicologie, cette partie vous sera
présentée dans le chapitre suivant « Evaluation de la toxicologie » ».
Les études de pharmacocinétique (Figure 7) permettent de décrire le comportement et le devenir du composé
dans un organisme vivant. Classiquement, il s'agit de modéliser son absorption, sa distribution dans l'organisme,
son métabolisme, et enfin à son élimination. Il s'agit des étapes ADME que vous avez abordé dans le cours
Monsieur Legeay.
Source : LEEM
10Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
Les études de toxicologie permettent de déterminer les effets potentiels toxiques et ainsi d'établir les organes
cibles et les doses toxiques du candidat médicament pour un organisme vivant. Nous allons nous attarder sur ces
études lors de ce cours évaluation de la toxicité).
Ces prérequis permettront de fixer les doses à administrer à l'Homme au cours des essais cliniques en appliquant
des marges de sécurité de manière à réduire au maximum les risques liés aux premières expositions humaines.
Les potentiels effets indésirables du futur médicament seront également déterminés, ce qui permettra de suivre
ces effets de manière proactive lors des essais cliniques.
Enfin, une partie du développement préclinique, appelée évaluation du risque environnemental, a pour objectif
de mesurer l'impact de la mise sur le marché d'un nouveau médicament pour l'environnement.
Source : LEEM
EVALUATION DE LA TOXICOLOGIE
« L’objectif des études de toxicologie préclinique est d'évaluer l'innocuité d'un candidat médicament
avant la phase clinique. Il faut imaginer que de nombreux candidats médicaments arrivent à cette étape du
développement mais arriveront-ils à la franchir pour arriver en phase clinique ? Quelques tests vont devoir être
passés. »
Pharmacologie de sécurité
Bien que faisant partie du dossier de pharmacologie, la pharmacologie de sécurité s'intéresse aux effets
éventuels sur les grandes fonctions vitales : système cardiovasculaire, système nerveux central et système
respiratoire (fréquence respiratoire, saturation...). Il faut imaginer qu'un candidat médicament ne passera pas la
future étape s'il met en danger ces fonctions vitales à doses pharmacologiques.
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
11Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Toxicité aiguë ou à dose unique
La toxicité aigüe vise à déterminer les doses toxiques chez l'animal et les organes souffrant effectivement
de cette toxicité.
Cette étape doit être réalisée chez au moins 2 espèces de mammifères (généralement rat et souris), avec 2 voies
d'administration (dont l'une sera la voie d'administration utilisée chez l'homme).
Le produit est administré à dose croissante. Chaque animal reçoit une dose unique de produit. La durée de suivi
après l'administration est généralement courte, de l'ordre de 14 jours. Cette étape permet de déterminer :
• La dose maximale tolérée : dose qui provoque un effet toxique mais qui n'affecte pas la survie des
animaux.
• La dose maximale sans effet toxique. Cette dose est appelée la NOAEL pour sa traduction en anglais : No
Observed Adverse Effect Level.
Toxicité chronique ou à doses réitérées
La toxicité chronique vise à obtenir des renseignements sur l'aptitude du produit à s'accumuler dans les
tissus et à confirmer quels organes souffrent sélectivement de cette toxicité.
Cette étape doit être réalisée chez au moins deux espèces de mammifères dont un non-rongeur (généralement
rat et chien/singe). La voie d'administration utilisée sera celle choisie pour l'administration humaine.
L'administration du produit est quotidienne ou bi-quotidienne. Lors de cette étape, 3 doses de produit sont
testées : une dose forte, une dose moyenne, une dose faible.
La durée recommandée dépend de la durée des essais cliniques futurs, elle-même gouvernée par les indications
futures chez l'Homme. En général on distingue les essais de toxicité subchronique qui durent de 1 à 3 mois et les
essais de toxicité chronique qui durent 6 mois.
Études de toxicité dans la reproduction et le développement
Avant-propos : les premières minutes de la vidéo qui suit vous expliquent très bien l'affaire du
thalidomide. Ce médicament sédatif, apparu à la fin des années 50, fut à l'origine d'une tragédie d'envergure
mondiale. Le thalidomide a notamment été prescrit à de nombreuses femmes enceintes afin de combattre la
nausée du matin. Il a été plus tard démontré qu'il était responsable de malformations fœtales irréversibles. Des
milliers d'enfants naquirent ainsi avec de graves malformations. Les effets indésirables sur l'embryon ou le fœtus
provoqués par des médicaments pris par la femme au cours de la grossesse sont appelés effets tératogènes.
Cette tragédie a permis de consolider les tests de reprotoxicité (toxicité de la reproduction). Les études de
reprotoxicité ont pour objectif d'évaluer les effets tératogènes, mais aussi l'impact du produit sur la fertilité et
ceux sur le développement post natal.
Ainsi, 3 types d'études sont réalisés :
- Fertilité et développement embryonnaire précoce jusqu'à implantation : réalisée sur une espèce de
rongeur, en général le rat. L'administration du produit a lieu plusieurs semaines avant l'accouplement sur
des mâles et des femelles. Leur capacité à se reproduire et à donner naissance à un embryon est observée
12Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
(nombre de spermatozoïdes, nidation, ...). Il ne serait pas admis qu'un médicament influence
négativement les capacités d'un couple à avoir des enfants.
- Développement embryo-fœtal/étude de tératogénèse : menée sur deux espèces depuis l'affaire du
thalidomide, un rongeur et un non-rongeur, généralement rat et lapin. Cette étude est réalisée sur des
femelles en gestation. L'administration du produit à lieu depuis l'accouplement jusqu'à la fin de
l'organogénèse. Les éventuelles malformations squelettiques, viscérales et morphologiques sont
observées.
- Développement pré et post-natal : réalisée sur une espèce de rongeur. Cette étude évalue l'impact du
produit sur la mise bas, le comportement maternel, l'allaitement et développement des petits. La
génération de petits peut être suivie sur du long terme et sa descendance également.
Ainsi le candidat médicament poursuivra son évaluation s'il ne présente, à priori, aucun risque de reprotoxicité,
c'est-à-dire, qu'il n'influence pas la fertilité, d'induit aucune malformation et permet un développement normal
de l'enfant (physique et mental).
Exemple : il est expliqué que l'isotrétinoïne (Curacné® induit des effets tératogènes. La prescription et la délivrance
de ce médicament traitant l'acné juvénile dont souffre la jeune Tartempion nécessite la prise d'une pilule
contraceptive et d'un suivi particulier en lien avec ses effets tératogènes (Encadré 3). Ici, le risque de malformations
par l'isotrétinoïne est important mais maitrisé car une contraception est mise en place chez cette jeune fille
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
Études de génotoxicité
L'évaluation de la génotoxicité correspond à l'ensemble des épreuves in vitro et in vivo par lesquelles il est
possible de détecter si un candidat médicament interagit avec l'ADN des cellules somatiques et/ou germinales et
qui, en l'absence de réparation fidèle, sont susceptibles de provoquer des mutations géniques et/ou
chromosomiques. Ces mutations géniques et chromosomiques sont susceptibles d'initier un processus
cancérogène lorsqu'elles ont lieu sur des cellules somatiques (sont toutes les cellules formant le corps d’un
organisme multicellulaire, c’est-à-dire toutes les cellules n’appartement pas à la lignée germinale, telles que
les gamètes, ou les cellules germinales). Les objectifs des différents tests de génotoxicité sont d'une part
d'identifier des agents génotoxiques (qui peut altérer l’information génétique codée par l’ADN), mais
également de déterminer leurs modes d'action moléculaires et cellulaires : bio-activation, nature des interactions
avec l'ADN, modifications de bases, cassures de brins, pontages, intercalations, substitutions, addition ou
délétion de paires de bases, cassures chromatidiennes ou chromosomiques, pertes de chromosomes entiers...
En effet, il existe un lien entre l'exposition à certains produits chimiques et la cancérogenèse chez l'humain et, les
épreuves de génotoxicité sont utilisées principalement pour prévoir la cancérogénicité.
13Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Autrement dit, il ne serait pas acceptable qu'un médicament soignant, par exemple, le diabète soit génotoxique
et donc potentiellement cancérogène. Un candidat médicament qui induirait des effets génotoxiques serait donc
éliminé, à cette étape. S'il s'agit d'un médicament destiné au traitement d'un cancer, il en sera peut-être
autrement en fonction du rapport bénéfice/risque.
Études de carcinogénicité
Une évaluation de la carcinogénicité (capacité à provoquer ou favoriser le cancer) d'un produit peut se
révéler nécessaire selon la durée du traitement et la population de patients visée.
Le cancer est une maladie provoquée par la transformation de cellules qui deviennent anormales et prolifèrent
de façon excessive. L'origine est souvent multifactorielle. En dehors des effets génétiques, l'exposition plus ou
moins chronique à un xénobiotique peut également être responsable d'effets cancérogènes. A titre d'exemple, il
est admis que le tabac favorise le développement du cancer pulmonaire et l'alcool celui du foie. Certains
médicaments, du fait d'une prise au long court, pourrait favoriser l'apparition de cancer, ou autrement dit être
cancérogène. Certains médicaments ont été retirés du marché car les études après sa mise sur le marché,
démontraient leur implication dans l'apparition de certains cancers. Parfois sans être retiré du marché, une
attention particulière est donnée comme celle avec l'hydrochlorothiazide que vous avez vu avec Monsieur
Legeay en pharmacologie qui serait lié à un risque de développer un cancer de la peau (lien vers l'information
sur le site de l'ANSM).
Les tests de cancérogenèse sont non requis avant la conduite d'un essai clinique sauf en cas de signes d'appels
comme l'analogie chimique avec des substances cancérogènes connues ou si des images histologiques suspectes
en toxicologie réitérée ont été obtenues.
Par exemple, les amines aromatiques sont connues pour induire des effets cancérogènes, il sera donc judicieux
de vérifier l'innocuité de ces molécules. De même qu'une image histologique de foie suspicieuse montrant des
cellules anormales et une prolifération cellulaire, induira des doutes qui devront être levés par une étude de
cancérogenèse.
En principe, l'étude de cancérogenèse se fait à long terme chez deux espèces différentes de rongeurs. La durée
de traitement est de 24 à 30 mois (rat) et 18 à 24 mois (souris) ce qui s'apparente à leur durée de vie entière. Ainsi,
cette exposition reproduira une exposition humaine chronique qui pourrait avoir lieu tout au long de la vie d'un
individu afin d'évaluer le risque encouru en cas de prise chronique d'un médicament.
3) Calcul de la première dose administrable à l’Homme
Un des objectifs des études précliniques est de déterminer la 1ère dose qui sera administrée à l'Homme
lors des études cliniques.
Ça y est ! Le candidat médicament a franchi les étapes clés de l'évaluation préclinique. Ce candidat médicament
semble être non toxique à court terme. Il est important de déterminer une dose initiale pour la nouvelle substance
active qui ne doit pas entraîner d'effets décelables à court terme.
14Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
Cette dose sera choisie sur la base de la NOAEL qui sera la dose de référence en matière de sécurité lorsqu'elle a
été établie à partir d'études appropriées sur des animaux. Comme, elle a été déterminée sur des animaux, un
calcul de conversion basée sur la surface corporelle est nécessaire pour transformer cette NOAEL en HED
pour Human equivalent doses. Il existe des grilles permettant de calculer cette dose en fonction de la masse de
l'espèce sur laquelle la molécule a été testée. Des facteurs seront différents en fonction de la pharmacocinétique
chez l'espèce animale et de la capacité de l'animal à être prédictif pour l'Homme. En général, un singe, se
rapprochant de l'Homme sera plus prédictif qu'une souris. Enfin, un facteur de sécurité doit être appliqué afin de
fournir une marge de sécurité pour la protection des sujets humains recevant la dose clinique initiale.
Ainsi la première dose maximale recommandée sera déterminée après ces étapes. Il s'agira de la MRSD
pour Maximum Recommended Starting Dose. Enfin, il sera judicieux de comparer cette dose à celle
pharmacologiquement active.
Voici un exemple :
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
4) Les études cliniques
Avant de pouvoir débuter un essai clinique en France, le protocole de l'étude doit être soumis à une
instance appelée le Comité de Protection des Personnes (CPP). Le CPP a pour rôle de s'assurer que le protocole
de l'étude respecte la sécurité des participants et la législation française et européenne en matière de recherche
clinique.
Composition d'un CPP : professionnels de santé, représentants de patients / juristes / psychologue / travailleur
social / spécialiste de l'éthique.
En parallèle, les promoteurs d'un essai doivent obligatoirement soumettre un dossier de demande d'autorisation
à l'Agence Nationale de Sécurité des Médicaments et des produits de santé (ANSM). Elle évalue la sécurité et la
qualité des produits utilisés au cours de la recherche.
Pour cela elle s'assure de :
- La qualité du produit (pureté/fabrication etc...)
- Les résultats des tests de toxicologie et s'ils ont été menées sous BPL.
- Les doses, NOAEL et la première dose administrable à l'Homme proposée.
Voici les 4 étapes de la recherche clinique (Figure 8). Ces étapes seront revues dans l'UE5 dans le cours du Dr
Briet.
15Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
Chaque produit pharmaceutique ayant obtenu une AMM possède une notice mais aussi des RCP pour « Résumés
des Caractéristiques du Produit ». L'ANSM publie sur son site les RCP et notices d'AMM demandées au niveau
national.
5) Pharmacovigilance
Une fois l'AMM obtenue, le médicament se retrouve sur le marché. Les médecins peuvent alors le
prescrire aux patients dans le respect de ses indications et les pharmaciens peuvent le dispenser en officine et/ou
à l'hôpital.
De nombreuses informations sont obtenues après la mise sur le marché car le médicament est délivré à une large
proportion de patients avec des caractéristiques physiopathologiques différentes (âge, origines ethniques,
comorbidité). Les effets indésirables non déterminés peuvent être transmis par tout professionnel de santé mais
également par les patients eux-mêmes aux services de pharmacovigilance ou à l'agence nationale du
médicament ou encore sur internet sur des sites dédiés.
Au fur et à mesure des déclarations plusieurs décisions peuvent être prises par l'ANSM. Cela permet parfois :
- De retirer du marché un médicament où le risque serait trop important par rapport au bénéfice (effet
thérapeutique) ;
- D'adapter la posologie ;
16Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
- De modifier les RCP présentes dans les boîtes de médicaments ;
- De modifier les modalités de délivrance du médicament.
C) Le paracétamol
VIDEO 1 : L’agence du médicament lance un avertissement concernant l’usage du paracétamol. Il ne faut pas en
prendre trop. C’est la substance active la plus consommé en France. Et la France est le plus gros consommateur
d’Europe. En 2018, la moitié de la population a eu une ordonnance pour le paracétamol, l’autre moitié le
consomme en vente libre. Il y a 500 millions de boites vendu par ans en France. L’ANSM vient donc de mettre sur
le marché de nouvelles boites avec le risque de toxicité indiqué sur la face avant. En fait, on peut faire une hépatite
(infection du foie) en cas de surdosage. Pour éviter ceci, il faut commencer par prendre la dose la plus faible, ne
pas dépasser 3g par jour, 1g par prise toute les 4h. Si le paracétamol n’est pas efficace, cela ne sert à rien
d’augmenter les doses, ce sera dangereux. Attention aux doses en cas de dénutrition, de poids faible,
d’insuffisance hépatique, attention aussi aux bébés. Pas de paracétamol après la gueule de bois (car l’alcool
attaque le foie aussi). Donc quand on se murge la gueule après l’examen, pas de paracétamol le lendemain !
VIDEO 2 : Cette vidéo montre que le paracétamol peut être toxique en cas de surdosage (inoffensif si posologie
normale). Si on surdose, le foie n’arrive plus à détruire le médicament. Il peut y avoir une hépatite fulminante. Le
foie est détruit (on peut en mourir). Il faut donc faire attention aux dosages !!
Historique :
A ce jour, il s'agit du médicament le plus prescrit et le plus vendu en pharmacie.
En France, il est délivré à l'hôpital ou en pharmacie de ville contrairement à certains pays anglo-saxons où le
paracétamol est en vente libre dans les supermarchés
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
Le paracétamol a été mis sur le marché en France en 1957. A cette époque, la phase d'évaluation du médicament
comme je vous l'ai décrite n'était pas en place. Cette molécule est largement utilisée pour son activité
antipyrétique (diminution de la fièvre) et son action antidouleur. Il s'agit du médicament le plus prescrit en France
mais également le plus vendu sans prescription auprès des pharmaciens.
17Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
La posologie indiquée sur les RCP est :
- La posologie unitaire usuelle est d'un comprimé à 1000 mg par prise, à renouveler au bout de 6 à 8 heures.
En cas de besoin, la prise peut être répétée au bout de 4 heures minimum.
- Il n'est généralement pas nécessaire de dépasser 3 g de paracétamol par jour, soit 3 comprimés par jour.
- Cependant, en cas de douleurs plus intenses, la posologie maximale peut être augmentée jusqu'à 4 g (4
comprimés) par jour. Toujours respecter un intervalle de 4 heures entre deux prises.
Une prise en une seule fois de 10 g de paracétamol chez l'adulte provoque une cytolyse hépatique susceptible
d'aboutir à une nécrose complète et irréversible se traduisant par une insuffisance hépatocellulaire, une acidose
métabolique, une encéphalopathie pouvant aller jusqu'au coma et la mort.
C'est pourquoi, les boite de paracétamol ne contiennent que 8 comprimés de 1000 mg car a fortiori, une
consommation de la boîte entière n'est pas mortelle (en théorie car elle peut conduire à une insuffisance
hépatique).
Nous pouvons trouver le paracétamol sous le nom de différentes spécialités : Doliprane®, Dafalgan®,
Efferalgan®... ou directement sous le nom de la dci (dénomination commune internationale) « paracétamol ». Il
n'est pas rare que les patients prennent ces spécialités en pensant qu'il s'agisse de molécules différentes. Il existe
également de nombreuses spécialités contenant du paracétamol et une autre molécule (Fervex Rhume ® à base
de paracétamol et chlorphénamine).
En 2008, les spécialités à base de paracétamol ont été mis en libre accès en officine, appelé aussi OTC pour Over
the counter, terme dérivé des pratiques anglo-saxonnes. Il était ainsi possible d'acheter du paracétamol en le
prenant directement dans le rayon de la pharmacie.
C'est ainsi que la consommation de paracétamol, largement utilisé dans notre quotidien pour la douleur ou en
cas de fièvre, peut être banalisée et conduire à de nombreuses intoxications.
Depuis janvier 2020, tous les médicaments à base de paracétamol se retrouvent derrière le comptoir nécessitant
ainsi de verbaliser la demande d'achat auprès de l'équipe officinale ou d'avoir une prescription. Vous verrez
également inscrit sur les boîtes de paracétamol ou de toute spécialité qui en contient qu'il s'agit bien d'un
médicament toxique pour le foie mais également qu'il faut se méfier de la prise d'autres médicaments contenant
du paracétamol (Figure 10).
Source : Cours « Le médicament en toute sécurité pour le patient » de Mme Savary
18Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
D) Conclusion
La découverte d'un médicament peut être fortuite ou le fruit de nombreuses années de recherche et
développement. Dans tous les cas, l'innocuité de chaque molécule doit être évaluée, les mécanismes de toxicité
étudiés et le rapport bénéfice/risque déterminé. Chaque patient doit pouvoir être serein quant aux produits
pharmaceutiques. Les 3 phases précliniques, clinique et de pharmacovigilance sont là pour s'en assurer.
19Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Récapitulatif
POINTS IMPORTANTS
I. Généralités
➢ Un médicament, bien qu’il soit sensé soigner, peut être toxique, il faut bien respecter les
doses et évaluer la notion de bénéfice/risque. Attention aux médicaments avec une marge
thérapeutique étroite !
➢ Attention aux médicaments avec une marge thérapeutique étroite !
II. L’évaluation de la sécurité du médicament lors de son cycle de vie
➢ Faire un médicament demande beaucoup de temps et d’argent. Il y a énormément de
molécules au début des essais pré-clinique. Leur nombre se réduit au fur et à mesure.
➢ Après avoir calculé, grâce aux essais précliniques, la première dose administrable chez
l’homme, on passe aux essais cliniques (en trois étapes !)
➢ Il faut garder en têt que le médicament est toujours suivi et surveillé même après sa mise sur
le marché. C’est la pharmacovigilance.
20Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
Entrainements
EXERCICE 1 :
Pour chaque situation, quelles les informations exactes ?
1- Dans la Situation 1 : la molécule A et B partagent le même bénéfice/risque : VRAI / FAUX
2- Dans la Situation 1 : pour une efficacité maximale, la molécule A n'engendrera pas d'effet toxique :
VRAI / FAUX
3- Dans la Situation 2 : la molécule B sera privilégiée car plus efficace : VRAI / FAUX
4- Dans la Situation 2 : la molécule B possède une très faible marge thérapeutique : VRAI / FAUX
5- Dans la Situation 3 : le médicament A sera privilégié car moins toxique malgré une meilleure efficacité de la
molécule B : VRAI / FAUX
6- Dans la Situation 4 : le médicament B sera privilégié car il engendre moins d'effets iatrogènes : VRAI / FAUX
EXERCICE 2 :
Quel est l’effet secondaire principal du paracétamol ?
A) Atteinte hépatique
B) Atteinte rénale
C) Atteinte cardiaque
21Faculté de Santé
UE4 : Sciences thérapeutiques Pr.Savary
Département PluriPASS
Correction
EXERCICE 1 :
1- FAUX
2- VRAI : A et B partage le même profil de toxicité mais l'efficacité de A est meilleur et surtout il n'engendre pas
d'effet toxique au doses thérapeutique.
3- FAUX Dans ce cas, la molécule A sera privilégié car la molécule B est très toxique. La molécule A possède un
bon rapport bénéfice risque car comporte peu de risque. C'est l'inverse pour la molécule B.
4- VRAI : C’est exact, voir même plus du tout de marge thérapeutique lorsqu’une certaine dose est dépassée
5- FAUX : Non la molécule B sera privilégiée car n'engendre pas d'effet toxique pour une efficacité maximale.
Le rapport bénéfice/risque est le même. Ces molécules possèdent la même marge thérapeutique
6- FAUX : Dans le cas d'une pathologie comme le cancer, nombreux médicaments engendrent des effets
secondaires que l'on dit « acceptables » car l’issue de la maladie est très grave. Ici les effets secondaires
seront tolérés au profit d'une molécule efficace.
EXERCICE 2 :
Réponse A (attention qcu)
22Faculté de Santé
UE4 : Sciences thérapeutiques Pr. Savary
Département PluriPASS
Notes
23Vous pouvez aussi lire

















































