Investigation des neuropathies de Charcot-Marie-Tooth
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
curriculum
Investigation des neuropathies de Charcot-Marie-Tooth
Susanne Renaud
Neurologische Klinik, Universitätsspital Basel, et Service de Neurologie, Hôpital neuchâtelois
mais impossible à confirmer catégoriquement. Une
étiologie inflammatoire à cette polyneuropathie semble
Quintessence
peu probable au vu des protéines totales normales dans
P Les neuropathies de Charcot-Marie-Tooth sont un groupe de polyneu- le liquide céphalorachidien. Une polyneuropathie dé-
ropathies héréditaires. myélinisante avec anti-MAG est exclue avec un titre
d’anticorps normal et une électrophorèse des protéines
P Il y a au moins 40 différentes mutations ayant différents modes de
normale.
transmission.
Il s’agit donc d’une polyneuropathie démyélinisante sur
P L’investigation rationnelle repose sur une anamnèse familiale fouillée, probable fond héréditaire (autosomale dominante) et à
la détermination du phénotype et l’électroneurographie, avant toute ana- début tardif (>50 ans). Comment peut-elle maintenant
lyse génétique. être attribuée à telle ou telle mutation?
Historique
Le cas
Pour avoir un aperçu des très nombreux géno- et
Un patient qui avait alors 60 ans est venu nous consul- phénotypes actuels des polyneuropathies héréditaires
ter pour la première fois en raison de douleurs dans et de leurs différentes classifications, il vaut la peine
son bras gauche et de brûlures occasionnelles dans les d’en présenter l’évolution historique.
doigts I–III de sa main gauche. Il signalait en outre des Depuis la première description en 1886 par Jean-Martin
fourmillements et douleurs de ses membres inférieurs Charcot et Pierre Marie d’une part et Howard Henry
avec insécurité à la marche, qu’il attribuait à une gon- Tooth de l’autre, le terme de neuropathies héréditaires
arthrose. L’anamnèse nous apprend que son frère aîné, de Charcot-Marie-Tooth (abrégé CMT) a été créé [1, 2].
maintenant décédé, de même que l’une de ses tantes et Dans leur description de ce syndrome d’«atrophie mus-
ses 4 filles avaient une démarche «bizarre». Ses pa- culaire péronière» familiale, ils avaient déjà précisé les
rents sont décédés jeunes, ce qui fait que nous n’avons caractéristiques majeures de cette maladie: une atrophie
aucune information sur leur manière de marcher. musculaire distale progressive avec faiblesse, déforma-
L’examen neurologique est frappant par des pieds tions squelettiques et hérédité. Jules Déjérine et Jules
creux très marqués, des réflexes ostéotendineux faibles Sottas ont décrit quelques années plus tard (1893) une
avec absence bilatérale du réflexe achilléen. Ce patient neuropathie beaucoup plus grave avec hypertrophie ner-
présente en outre une hypoesthésie en socquette à tous veuse chez deux enfants d’une même famille [3].
les examens avec pallesthésie au niveau des orteils et C’est en 1957 que Gilliat et Thomas [4] et plus tard Dyck
baisse de la perception spatiale. Le Romberg et la mar- et Lambert (1968) ont décrit pour la première fois un
che en ligne droite sont hésitants. ralentissement massif de la vitesse de conduction ner-
Nous effectuons en premier lieu une électroneurogra- veuse dans quelques familles présentant une neuropa-
phie du n. médian dans l’optique d’un syndrome du thie héréditaire, alors que dans d’autres elle était pra-
tunnel carpien. Le temps de conduction au niveau du tiquement normale [5, 6]. C’est ceci qui a permis de
poignet est nettement prolongé non seulement pour faire la distinction entre les formes démyélinisante et
le n. médian mais aussi pour le n. cubital et les vitesses axonale des neuropathies héréditaires. Dyck et Lam-
de conduction motrice sont légèrement ralenties pour bert ont ensuite introduit une classification des neuro-
ces deux nerfs au niveau de l’avant-bras. Les vitesses pathies sensitivomotrices héréditaires (HMSN) qui
de conduction sensitive sont nettement diminuées pour porte leurs noms, selon laquelle le type I est le type dé-
le n. médian des deux côtés et juste dans les normes in- myélinisant autosomal dominant et le type II le type
férieures pour le n. cubital. A l’électroneurographie des axonal autosomal dominant. Anita Harding et P. K. Tho-
Susanne Renaud membres inférieurs, nous sommes frappés par une mas ont observé en 1980 une distribution bimodale des
polyneuropathie sensitivomotrice très marquée avec si- vitesses de conduction nerveuse dans le n. médian de
gnes d’une lésion aussi bien démyélinisante qu’axonale. plusieurs familles présentant des neuropathies hérédi-
L’auteur certifie Les vitesses de conduction motrice sont fortement ra- taires et ont fixé comme limite de distinction entre
qu’aucun conflit lenties (par ex. pour le n. péronier gauche 23 m/s et le HMSN I et II une vitesse de 38 m/s [7].
d’intérêt n’est
n. tibial 17 m/s avec des amplitudes globalement fai- En 1992, plusieurs groupes de recherche indépendants
lié à cet article.
bles). Un syndrome du tunnel carpien est probable, ont montré que l’étiologie de la maladie de Charcot-Ma-
Vous trouverez les questions à choix multiple concernant cet article
à la page 434 ou sur Internet sous www.smf-cme.ch. Forum Med Suisse 2010;10(25):441–444 441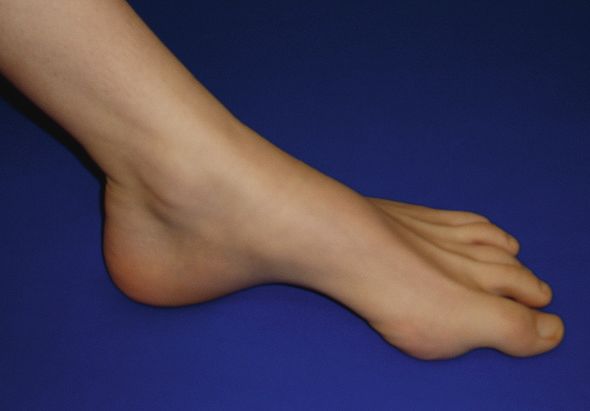
curriculum
à 17–40/100 000 [12]. Son phénotype est généralement
Tableau 1. Classification.
homogène avec atrophies et faiblesse de la musculature
CMT Description Dyck/Lambert distale des membres, la musculature péronière étant la
CMT 1 A–F Autosomale dominante démyélinisante HMSN I plus fortement touchée, la plupart du temps avec trou-
HNPP bles symétriques de la sensibilité distale, déformations
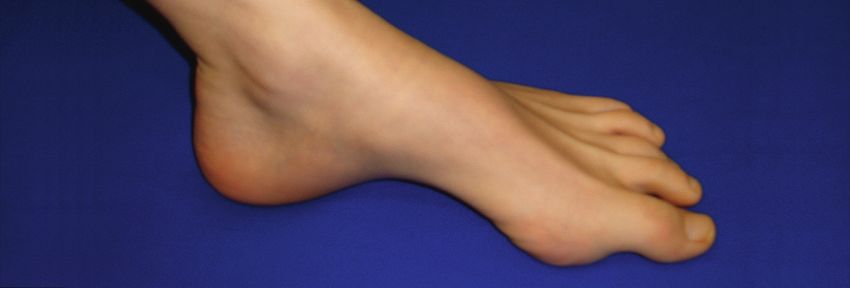
CMT 2 A–L Autosomale dominante axonale HMSN II squelettiques (pied creux, voir fig. 1 x) et réflexes os-
DI-CMT A–D Dominante type intermédiaire
téotendineux faibles ou absents. C’est pour cette raison
que les différentes formes génétiques de CMT sont pra-
CMT X Liée au sexe tiquement impossibles à distinguer chez un seul pa-
dominante démyélinisante
tient, et imposent un engagement complexe avec mode
CMTX 2–5 Liée au sexe de transmission héréditaire et examen électrophysio-
récessive axonale
logique. La gravité de cette maladie peut même être très
CMT 3 A–D Gravement démyélinisante HMSN III variable au sein d’une même famille.
Déjérine-Sottas à début précoce
AR et AD
CMT 4 A–J Autosomale récessive démyélinisante
Mode de transmission
CMT4 C1 ou AT-CMT2 Autosomale récessive axonale
Des formes autosomales dominantes et liées au sexe
Maladie de Refsum HMSN IV
ont été décrites. Les formes démyélinisantes autosoma-
CMT avec signes pyramidaux Dominante axonale HMSN V les récessives (CMT4) commencent pratiquement tou-
Avec atrophie du nerf optique, HMSN VI jours tôt et ont une évolution plus grave que les formes
dominante axonale dominantes. Les formes axonales autosomales récessi-
dHMN I–VII Distal hereditary motor neuropathy ves sont très rares [13].
HSAN I–IV Hereditary Sensory Autonomic
Neuropathies, AD et AR et liée au sexe
Electroneurographie
L’examen électrophysiologique permet d’évaluer la fonc-
tion des fibres nerveuses motrices et sensitives. Un
fort ralentissement de la vitesse de conduction ner-
veuse (VCN) parle pour une affection de la gaine de
myéline et une diminution d’amplitude des potentiels
d’action moteurs globaux pour une atteinte d’abord
axonale. Il va de soi que des exceptions ont été décrites.
Il y a par ex. le groupe lié au sexe, dû à une mutation
du gène GJB1, avec une VCN intermédiaire [14]. Il y a
aussi des CMT autosomales dominantes avec VCN in-
termédiaires entre essentiellement axonales et essen-
tiellement démyélinisantes [15].
L’une des principales fonctions de l’électrophysiologie
est de faire la distinction par rapport à d’autres neuro-
pathies essentiellement démyélinisantes inflamma-
toires et de préciser exactement dans quelle mesures
les nerfs moteurs ou sensitifs sont atteints.
Figure 1
Pied creux chez une patiente ayant une neuropathie héréditaire (HNPP)
avec tendance aux parésies à la pression.
Différents gènes
rie-Tooth 1A était une duplication dans le gène PMP22 Nous connaissons maintenant au moins 40 dif-
[8–11]. Depuis le début de l’ère de la biologie molécu- férentes formes de CMT (www.molgen.ua.ac.be et
laire, nous reparlons de CMT et plus de HMSN. Le terme www.neuro.wustl.edu). Nous ne décrirons ici que les
de CMT3 n’est jamais parvenu à s’imposer et nous par- gènes les plus importants et leurs produits.
lons ici de neuropathie de Déjérine-Sottas (DSN). La dé-
couverte de très nombreux gènes avec des phénotypes PMP22
en partie identiques a donné lieu à une adaptation per- Le PMP22 est un composant de la myéline compacte,
manente de la classification (tab. 1 p). nécessaire pour la myélinisation correcte et son entre-
tien dans les nerfs périphériques. La duplication hété-
rozygote du segment contenant le gène PMP22 sur le
Epidémiologie et clinique chromosome 17 est associée à la forme la plus fré-
quente de CMT, la CMT1A. Une délétion provoque la
La maladie de CMT est la pathologie neuromusculaire neuropathie héréditaire avec tendance aux parésies à
héréditaire la plus fréquente, sa prévalence est estimée la pression (HNPP) [16]. Les mutations «missense» du
Forum Med Suisse 2010;10(25):441–444 442curriculum
Tableau 2. Stratégie d’investigation des CMT (d’après Szigeti 2006).
Electrophysiologie
Hérédité Démyélinisante Intermédiaire Axonale
Autosomale dominante Duplication PMP22 70% DNM MFN2 20%
Mutation MPZ 5% MPZ MPZ
Mutation PMP22 2,5%
Autres: LITAF, NEFL, EGR2 Autres: RAB7, GARS, NEFL, HSP27, HSP22
Autosomale récessive Rares: PRX, GDAP1, EGR2, MTMR2, Rares: GDAP1, LMNA, TDP1
SBF2, NDRG1, SH3TC2
Liée au sexe GJB1 12% GJB1 12% GJB1 12%
Abréviations: DNM2 = dynamine 2, EGR2 = early growth response 2, GARS = glycil-tRNA synthetase, GDAP1 = gangliosid-induced
differentiation-associated protein 1, GJB1 = gap-unction B1/connexine 32, HSP27 = heat-shock protein 27-kDa protein 1, HSP22 =
heat-shock 22-kDa protein 8, LITAF = lipopolysaccharide-induced tumor necrosis factor-a, MPZ = myelin protein zero, MTMR2 = myotubula-
rin-related protein – 2, NDRG1 = Nyc downstream-regulated gene 1, NEFL = neurofilament light chain, PMP22 = peripheral myelin protein 22,
PRX = periaxin, SBF2 = set-binding factor 2, SH3TC2 = SH3 domain and tetratricopeptide repeat domain 2.
PMP22 provoquent les phénotypes de Déjérine-Sottas, superficiels qui sont touchés dans la CMT2A [21]. Mais
plus sévères. Les mécanismes cellulaires exacts de la rares sont les travaux qui ont été publiés à ce sujet, et
pathogenèse ne sont cependant pas connus (aperçu nous devons encore attendre pour savoir quelle est la
dans [17]). spécificité de l’IRM musculaire à différents stades de
ces maladies.
Myelin Protein Zero (MPZ, P0)
La P0 est une protéine transmembranaire avec un
domaine immunoglobulinoïde, elle totalise env. 50% Investigation rationnelle en cas de suspicion
des protéines de la myéline périphérique. Elle sert de de polyneuropathie héréditaire
molécule d’adhésion entre les couches adjacentes de
myéline. Fait intéressant, les mutations de la MPZ don- Pour une investigation rationnelle, il faut commencer
nent plusieurs phénotypes. Un groupe a un début très par préciser le phénotype clinique avec l’âge du début
précoce (première décennie) de la maladie et des VCN de la maladie et classer la vitesse de conduction ner-
très lentes, alors que l’autre a un début plus tardif avec veuse en axonale, démyélinisante ou intermédiaire
dégénérescence axonale, et très peu de patients seule- (tab. 2 p). Le mode de transmission doit être analysé
ment ont le phénotype électromyographique classique le plus précisément possible. Le problème génique le
de la CMT1A [18]. plus fréquent dans la CMT démyélinisante, à savoir la
CMT1, est à 70% une duplication du gène PMP22. En-
Mitofusine 2 (MFN2) suite vient une mutation du GJB1 dans la CMTX liée au
La MFN2 est une GTPase transmembranaire mitochon- sexe avec 12,5%, suivie des mutations de la MPZ (5%)
driale et un important élément de régulation de la fu- et du PMP22 (2,5%). Dans les très rares formes démyé-
sion mitochondriale. Les mutations donnent d’une part linisantes héréditaires autosomales récessives, toute
une perturbation de la physiologie des mitochondries et une série de défauts géniques entrent en ligne de
de l’autre un transport axonal mitochondrial patholo- compte, qui peuvent en partie être rapprochés du
gique. Elles sont responsables de 11 à 20% des polyneu- phénotype élargi. La plupart de ces gènes ne peuvent
ropathies axonales autosomales dominantes [17, 19, 20]. cependant pas être analysés commercialement en
Nous connaissons ici aussi des phénotypes plus ou Suisse, ce qui doit par conséquent se faire en collabo-
moins gravement atteints. ration avec des centres de recherche spécialisés. C’est
le plus souvent une mutation du GJB1 qui est trouvée
Phénotype élargi avec des vitesses de conduction nerveuse intermé-
Bien que de nombreuses mutations génétiques donnent diaires. Si le mode de transmission est autosomal domi-
des phénotypes pratiquement identiques, certains élé- nant, il faut penser à une mutation du gène de la MPZ
ments cliniques peuvent être utiles. Par exemple une ou de la dynamine. Bien que de très nombreuses mu-
atrophie du nerf optique, pouvant témoigner d’une mu- tations aient été décrites dans les polyneuropathies
tation de la MFN2 en relation avec une polyneuropathie axonales héréditaires, il semble que les plus fréquentes
axonale autosomale dominante, ou la manifestation de soient dues à des mutations du gène MFN2 (env. 20%).
signes pyramidaux subcliniques dans les neuropathies Il est en outre possible d’analyser les gènes MPZ et
CMTX [13]. Avec une IMR de la musculature, nous ten- GJB1 [22, 23].
tons de démontrer l’atteinte échelonnée dans le temps Il va de soi qu’une discussion approfondie doit avoir
de groupes musculaires bien précis. Alors que dans la lieu avant la prise de sang, éventuellement avec
CMT1A par ex. c’est surtout la musculature péronière l’assistance d’un spécialiste en génétique humaine. Le
qui est atteinte, ce sont plutôt les muscles postérieurs consentement éclairé écrit et signé est important.
Forum Med Suisse 2010;10(25):441–444 443curriculum
Traitement Correspondance:
PD Dresse S. Renaud
Il n’y a actuellement aucun traitement causal efficace Médecin-cheffe
Service de neurologie
des polyneuropathies de CMT. Du fait que l’expéri-
Département médecine
mentation animale a montré un effet positif de l’acide
Hôpital neuchâtelois
ascorbique sur la survie de rats porteurs d’une duplica- CH-2007 Neuchâtel
tion PMP22, une grande étude multicentrique est en susanne.renaud@ne.ch
cours avec cette substance. Il est probable qu’elle joue
un rôle dans le programme de myélinisation des nerfs
Références recommandées
périphériques [24, 25]. – Pareyson D, Scaioli V, Laurà M. Clinical and electrophysiological as-
pects of Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;
8(1-2):3–22.
– Nicholson G, Nash J. Intermediate nerve conduction velocities define
Solution du cas X-linked Charcot-Marie-Tooth neuropathy families. Neurology. 1993;
43(12):2558–64.
Une mutation «missense» du gène MPZ a finalement – Shy ME, Jáni A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Pheno-
typic clustering in MPZ mutations. Brain. 2004;127(Pt 2):371–84.
été découverte chez notre patient. Ce qui a permis de – Verhoeven K, Claeys KG, Züchner S, Schröder JM, Weis J, Ceuterick
poser le diagnostic de CMT1B. Sont parfaitement com- C, et al. MFN2 mutation distribution and genotype/phenotype corre-
patibles avec ce diagnostic l’hérédité autosomale domi- lation in Charcot-Marie-Tooth type 2. Brain. 2006;129(Pt 8):2093–102.
– Szigeti K, Nelis E, Lupski JR. Molecular diagnostics of Charcot-Marie-
nante, le début tardif et une atteinte axonale évidente à Tooth disease and related peripheral neuropathies. Neuromolecular
l’électroneurographie. Med. 2006;8(1-2):243–54.
Vous trouverez la liste complète et numérotée des références dans la
version en ligne de cet article sous www.medicalforum.ch.
Conclusion
Une anamnèse fouillée, la recherche du mode de trans-
mission et la caractérisation du phénotype permettent
de faire une première classification des CMT. Les possi-
bilités de la biologie moléculaire toujours plus nom-
breuses offrent ensuite une investigation rationnelle.
Forum Med Suisse 2010;10(25):441–444 444Die Abklärung der Charcot-Marie-Tooth-Neuropathien /
Investigation des neuropathies de Charcot-Marie-Tooth
Literatur (Online-Version) / Références (online version)
1 Charcot JM, Maire P. Sur une forme pariculière d’atrophie musculaire progressive
souvent familial debutant par les pieds et les jambes et atteignant plus tard les mains.
Rev Med. (Paris) 1886;6:97–138.
2 Tooth HH. The peroneal type of progressive muscular atrophy. London: HK Lewis;
1886.
3 Déjérine J, Sottas J. Sur la névrite: interstitielle, hypertrophique et progressive de
l’enfance. CR Sco Biol. (Paris). 1893;45:63–96.
4 Gilliatt RW, Thomas PK. Extreme slowing of nerve conduction in peroneal muscular
atrophy. Ann Phys Med. 1957;4:104–6.
5 Dyck PJ, Lambert EH. Lower motor and primary sensory neuron disease with
peroneal muscular atrophy I. Neurologic, genetic and electrophysiologic findings in
hereditary polyneuropathies. Arch Neurol. 1968;18:603–18.
6 Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with
peroneal muscular atrophy. II. Neurologic, genetic and electrophysiologic findings in
various neuronal degenerations. Arch Neurol. 1968;18:619–25.
7 Harding AE, Thomas PK. The clinical features of hereditary motor and sensory
neuropathy types I and II. Brain. 1980;103:259–80.
8 Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask BJ, Pentao L, et al. The gene
for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth
disease type 1A. Nat Genet. 1992;1(3):159–65.
9 Timmerman V, Nelis E, Van Hul W, Nieuwenhuijsen BW, Chen KL, Wang S, et al.
The peripheral myelin protein gene PMP-22 is contained within the Charcot-Marie-
Tooth disease type 1A duplication. Nat Genet. 1992;1(3):171–5.
10 Valentijn LJ, Bolhuis PA, Zorn I, Hoogendijk JE, van den Bosch N, Hensels GW, et
al. The peripheral myelin gene PMP-22/GAS-3 is duplicated in Charcot-Marie-Tooth
disease type 1A. Nat Genet. 1992;1(3):166–70.
11 Matsunami N, Smith B, Ballard L, Lensch MW, Robertson M, Albertsen H, et al.
Peripheral myelin protein-22 gene maps in the duplication in chromosome 17p11.2
associated with Charcot-Marie-Tooth 1A. Nat Genet. 1992;1(3):176–9.
12 Martyn CN, Hughes RA. Epidemiology of peripheral neuropathy. J Neurol Neurosurg
Psychiatry. 1997;62(4):310–8.
13 Pareyson D, Scaioli V, Laurà M. Clinical and electrophysiological aspects of Charcot-
Marie-Tooth disease. Neuromolecular Med. 2006;8(1-2):3–22.
14 Nicholson G, Nash J. Intermediate nerve conduction velocities define X-linked
Charcot-Marie-Tooth neuropathy families. Neurology. 1993;43(12):2558–64.
15 Verhoeven K, Villanova M, Rossi A, Malandrini A, De Jonghe P, Timmerman V.
Localization of the gene for the intermediate form of Charcot-Marie-Tooth to
chromosome 10q24.1-q25.1. Am J Hum Genet. 2001;69(4):889–94.
16 Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, et al.
DNA deletion associated with hereditary neuropathy with liability to pressure palsies.
Cell. 1993;72(1):143–51.
17 Niemann A, Berger P, Suter U. Pathomechanisms of mutant proteins in Charcot-
Marie-Tooth disease. Neuromolecular Med. 2006;8(1-2):217–42.
18 Shy ME, Jáni A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Phenotypic clustering
in MPZ mutations. Brain. 2004;127(Pt 2):371–84.2
19 Verhoeven K, Claeys KG, Züchner S, Schröder JM, Weis J, Ceuterick C, et al. MFN2
mutation distribution and genotype/phenotype correlation in Charcot-Marie-Tooth
type 2. Brain. 2006;129(Pt 8):2093–102.
20 Züchner S, De Jonghe P, Jordanova A, Claeys KG, Guergueltcheva V, Cherninkova S,
et al. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2.
Ann Neurol. 2006;59(2):276–81.
21 Chung KW, Suh BC, Shy ME, Cho SY, Yoo JH, Park SW, et al. Different clinical and
magnetic resonance imaging features between Charcot-Marie-Tooth disease type 1A
and 2A. Neuromuscul Disord. 2008;18(8):610–8.
22 Szigeti K, Nelis E, Lupski JR. Molecular diagnostics of Charcot-Marie-Tooth disease
and related peripheral neuropathies. Neuromolecular Med. 2006;8(1-2):243–54.
23 Jani-Acsadi A, Krajewski K, Shy ME. Charcot-Marie-Tooth neuropathies: diagnosis
and management. Semin Neurol. 2008;28(2):185–94.
24 Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, et al.
Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-
Tooth disease. Nat Med. 2004;10(4):396–401.
25 Pareyson D, Schenone A, Fabrizi GM, Santoro L, Padua L, Quattrone A, et al. CMT-
TRIAAL Group. A multicenter, randomized, double-blind, placebo-controlled trial of
long-term ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A (CMT-
TRIAAL): the study protocol [EudraCT no.: 2006-000032-27] Pharmacol Res.
2006;54(6):436–41.Vous pouvez aussi lire

















































