Les Insuffisances Surrénaliennes - Vendredi 23 Juin 2017 - Paris Programme et résumés - Association Surrénales
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
5e Journée de l’Association Surrénales
Les Insuffisances
Surrénaliennes
En partenariat avec les Centres de Référence labellisés
pour ces maladies, la filière Firendo.
Programme et résumés
des communications
Program and Abstracts
Vendredi 23 Juin 2017 - Paris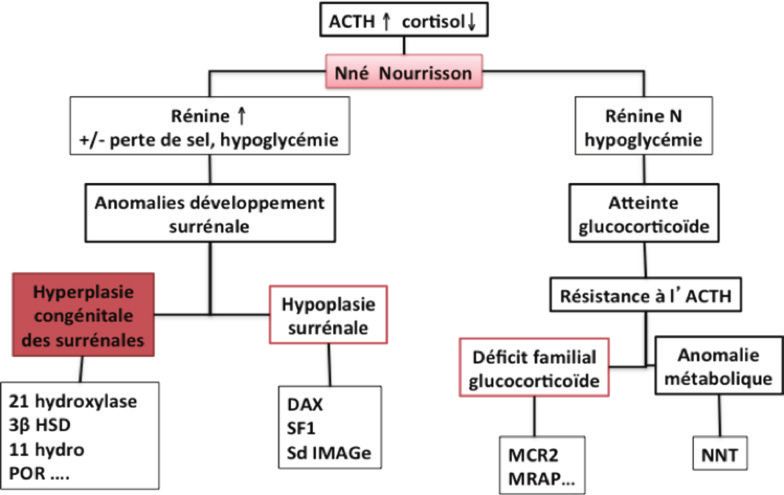
18:35 Page1
29/03/2017
CS_1_2013
visuel.qxp_H
Assoc_surrn_
ssociati on Surrénales
5 Journée de l’A
e
Les In su ffi sa nce s
Su rré na lie nn es
ellisés
c le s C e n tre s d e Référence lab
ave
En partenariat ce endo.
ies, la filière Fir
pour s malad
g ra m m e e t résumés
Pro n ications
de s co m m u
r a m a n d A b stracts
Prog
2017 - 06
uin 2 017 - Paris
78 18 - ©Paris
Vendredi 2 3 J
Plus - 06 08 32
e : La Com et en pag
Création et mise
www.surrenales.com
1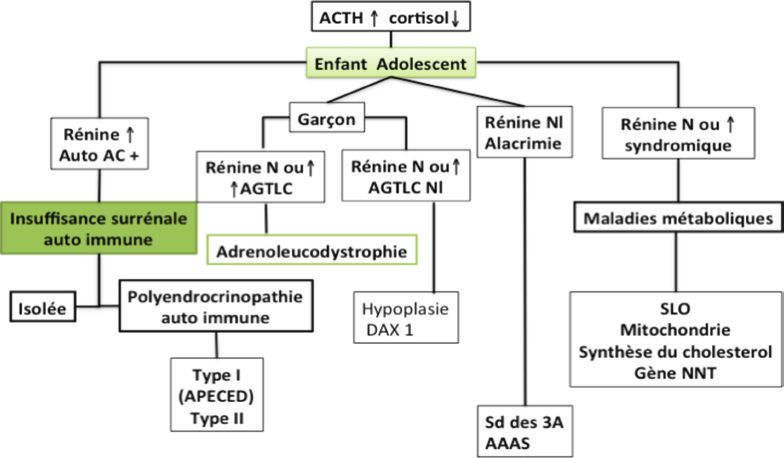
Conseil Scientifique
Présidé par le Pr Philippe Touraine Endocrinologue Pitié Salpêtrière Paris
Pr Yves Reznik Endocrinologue CHU Caen président de la journée thématique
Dr Claire Bouvattier Pédiatre endocrinologue Hôpital Bicêtre Kremlin Bicêtre
Dr Lise Duranteau Gynécologue endocrinologue Hôpital Bicêtre Kremlin Bicêtre
Dr Véronique Tardy Médecin biologiste CHU Lyon
Dr Delphine Zenaty Pédiatre endocrinologue Hôpital Robert Debré Paris
Pr Jérôme Bertherat Endocrinologue Hôpital Cochin Paris
Pr Pierre Mouriquand Chirurgien pédiatrique CHU Lyon
Pr Michel Polak Pédiatre endocrinologue Hôpital Necker-Enfants Malades Paris
Pr Jacques Young Endocrinologue Hôpital Bicêtre Kremlin Bicêtre
Les Insuffisances Surrénaliennes - 23 Juin 2017 2
Programme
8h30 : Accueil des participants 14h-14h30 : Les «take-home messages» du
Consensus Français sur le prise en charge
de l’insuffisance surrénale
9h00 - 9H10 : Introduction Yves Reznik, Caen
Claudine Colin et Philippe Touraine
Modérateurs : Rachel Reynaud et Jérôme Bertherat
1ère table ronde : Insuffisance surrénale
de l’enfant
Modérateurs : Michel Polak et Lise Duranteau 2e table ronde : Insuffisance surrénale
de l’Adulte
Modérateurs : Catherine Pienkowski et Hervé Lefebvre
9h10-9h30 : Cas clinique d’insuffisance
surrénale chez l’enfant
Delphine Zenaty, Paris 14h30-14h50 : Cas clinique d’insuffisance
9h30-10h : Comment orienter chez l’enfant
surrénale chez l’adulte
Delphine Vezzosi, Toulouse
le bilan étiologique d’une insuffisance
surrénale ? 14h50-15h15 : Morbidité de l’insuffisance
Dinane Samara-Boustani, Paris surrénale chez les patients hospitalisés
Gudmundur Johannsson, Gothenburg, Suède
10h-10h30 : Génétique de l’insuffisance
surrénale primaire congénitale 15h15-15h40 : Insuffisance surrénale
Florence Roucher-Boulez, Lyon et grossesse
Frédéric Castinetti, Marseille
10h30-11h : Insuffisance surrénale aiguë de
l’enfant
Claire Bouvattier, Paris
15h40 - 16H10 : Pause
11h00 - 11H30 : Pause 16h10-16h35 : Infusion continue par pompe
ou formes modifiées de l’hydrocortisone :
11h30-12h : La transition chez l’enfant
Laquelle choisir ?
Mariane Øksnès, Bergen, Norvège
insuffisant surrénalien
Philippe Touraine, Paris 16h35-17h : Les outils de l’éducation
thérapeutique du patient addisonien
Laurence Guignat, Paris
12h00 - 12H45 : Conférence Plénière : 17h-17h15 : Insuffisances surrénales : le
Insuffisance surrénale des états critiques point de vue des patients
Greet Van den Berghe, Louvain, Belgique Claudine colin et Christine Rougeau
Modérateurs : Muriel Houang et Anne Bachelot
17h30 : Conclusion & Synthèse
12h45 - 14H : Déjeuner Juliane Léger et Sophie Christin-Maitre
3
1 - CAS CLINIQUE D’INSUFFISANCE SURRENALE DE L’ENFANT :
NON DISPONIBLE
Delphine Zenaty, service Endocrinologie, Diabètologie Pédiatrique, Hôpital Robert Debré, APHP
Centre de Référence des Maladies Endocriniennes Rares de la Croissance et du développement
Notes
Les Insuffisances Surrénaliennes - 23 Juin 2017 4
Notes
5
2 - COMMENT ORIENTER CHEZ L’ENFANT LE BILAN ETIOLOGIQUE D’UNE INSUFFISANCE
SURRENALE?
Dinane Samara-Boustani. Service d’Endocrinologie, Diabétologie et Gynécologie Pédiatriques. Hôpital
Necker Enfants-Malades. Centre de Référence des Maladies Endocriniennes Rares de la Croissance et
du développement
Les étiologies de l’insuffisance surrénalienne (IS) de l’enfant sont essentiellement congénitales d’origine gé-
nétique. Elles se divisent en 4 groupes: 1. Les anomalies de la stéroïdogénèse 2. La dysgénésie / l’hypoplasie
surrénalienne 3. La résistance à l’ACTH et 4. La destruction du parenchyme surrénalien (Malikova 2014)
L’etiologie dépend de l’âge avec une prédominance des causes génétiques en période néonatale puis autoim-
mune et acquises dans l’enfance.
Selon l’âge de l’enfant et l’association ou non à un deficit mineralocorticoïde (associant hyponatrémie, hyperka-
liémie, taux de rénine augmenté), l’orientation étiologique diffère.
En période néonatale, l’étiologie prédominante est l’hyperplasie congénitale des surrénales (HCS) par déficit en-
zymatique de la stéroidogénèse, le plus fréquent étant le déficit en 21 hydroxylase; elle représente 45 à 60% des
cas (Leblicq 2011, Hsieh 2011). L’HCS se manifeste en période néonatale par une anomalie des organes génitaux
de la fille et/ou du garçon selon le bloc enzymatique. Le dépistage du deficit en 21 hydroxylase par le dosage de la
17OHP au buvard à J3 de vie permet d’éviter et de traiter rapidement les nouveaux nés atteints (avant ou au début
du syndrome de perte de sel par deficit en minéraocorticoïdes et les hypoglycémies sévères).
L’hypoplasie surrénalienne est la 2ème cause génétique et représente 1.5 à 5% selon les séries (hors HCS) (Perry
2005, Hsieh 2011). L’hypoplasie surrénalienne est due en premier lieu aux mutations du gène DAX 1. L’insuffisance
surrenalienne (gluco et minéralocorticoïde) survient le plus souvent dans le 1er mois de vie, mais il existe des
cas décrits dans l’enfance (Guran 2016). L’atteinte surrénalienne peut être associée à un hypogonadisme hypo-
gonadotrope d’intensité variable. La délétion du gène DAX1 peut s’intégrer dans le syndrome des gènes contigus
avec l’association de la maladie de Duchenne et d’un déficit en glycerol kinase. Les mutations du gène SF1,
responsables de dysgénésie gonadique, peuvent être responsables d’IS de manière épisodique (quelques cas dé-
crits dans la littérature). L’hypoplasie surrénalienne peut également s’intégrer dans d’autres syndromes comme:
IMAGe syndrome (CDKN1C), Pallister-Hall syndrome (GLI 3) , Meckel syndrome, Pena-Shokeir syndrome (DOK7,
RAPSN) , Pseudotrisomy 13 ,Hydrolethalus syndrome (HYLS1) (Malikova 2014).
La résistance à l’ACTH représente 9% des IS (Hsieh 2011, Tsai 2016). Cela engloble: le déficit en glucocorticoides
familial (mutation MC2R et MRAP) se manifestant par des hypoglycémies néonatales, des convulsions, une ano-
malie du développement psychomoteur et une mélanodermie; le Triple A syndrome associant une alacrimie dès
la naissance, une achalasie, une surdité, une atteinte neurologique progressive et un déficit en glucocorticoïdes
qui survient entre l’âge de 5 et 10 ans; les anomalies du stress oxydatif par mutation du gène NNT récemment
décrit (Meimaridou 2012), l’IS est diagnostiquée dans les 2 premières années de vie mais parfois plus tard (Rou-
chez-Boulez 2016).
Dans l’enfance, l’IS peut être due à une destruction du parenchyme surrénalien. La première cause à rechercher
est l’insuffisance surrénalienne auto immune par le dosage des anticorps anti 21 hydroxylase. Elle représente 21
à 43% des IS avec une moyenne d’âge d’apparition des signes entre 11 et 14 ans. L’IS auto immune peut s’intégrer
dans une polyendrocrinopathie auto immune ou syndrome d’APECED type 1 lié à des mutations du gène AIRE qui
représente 8 à 12% des IS. Le diagnostic est fait sur l’association de 2 des 3 critères qui sont la candidose cutanéo
muqueuse (apparaissant dans l’enfance), l’hypoparathyroïdie (apparaissant vers l’âge de 7 ans) et l’IS survenant
entre l’âge de 5 et 15 ans avec une augmentation progressive de la prévalence avec l’âge (Perheentupa 2006). En
cas d’absence d’anticorps ou en cas de signes neurologiques associés, Il faut rechercher, chez le garcon, une
adrenoleucodystrophie (ALD) par le dosages des acides gras à très longues chaines. Cette pathologie est liée à
une mutation du gene ABCD1. L’ALD représente 7% des IS (hors HCS ) (Hsieh 2011, Perry 2005) et 11 à 30% des IS
des garçons (Hsieh 2011, Perry 2005, Simm 2014). L’IS apparaît en moyenne entre l’âge de 10 et 12 ans.
D’autres pathologies peuvent être responsables de la destruction du parenchyme surrénalien comme le syn-
drome de Zellweger (PEX genes), la maladie de Refsum (genes PHYH, PEX7), les cytopathies mitochondriales
(Kearne Sayre syndrome, MELAS syndrome); les anomalies de la synthèse du cholesteriol (syndrome de Wol-
man, syndrome de Smith Lemly opitz)
Les causes acquises d’IS sont beaucoup plus rares :
Les Insuffisances Surrénaliennes - 23 Juin 2017 6
Les hémorragies surrénaliennes, les infections sévères (choc septique, HIV, tuberculose, CMV), la chirurgie, les
traumatismes, les pathologies infiltratives (métastase, lymphome, sarcoïdose, hemochromatose) et les medica-
ments (ketoconazole, rifampycine, phenytoïne)
L’orientation étiologique de l’IS de l’enfant doit se faire en fonction de l’âge, de la presence ou non d’une atteinte
minéralocorticoïdeet des signes cliniques associés qu’il faut savoir rechercher. Il existe une prédominance des
causes génétiques en période néonatale (HCS) et auto immunes chez l’enfant plus grand. L’adrenoleucodystro-
phie doit être évoquée chez tous les garcons avec une IS à anticorps anti 21 hydroxylase négatifs.
Orientation étiologique de l’IS en période néona-
tale
Orientation étiologique de l’IC chez l’enfant
Bibliographie:
- Guran T. et al. Rare Causes of Primary Adrenal Insufficiency: Genetic and Clinical Characterization of a Large Nationwide Cohort. J
Clin Endocrinol Metab 101: 284 –292, 2016
- Hsieh S. et al. Presentation of Primary Adrenal Insufficiency in Childhood. J Clin Endocrinol Metab 96: E925–E928. 2011
- Husebye E. S. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. Journal of
Internal Medicine 265; 514–529, 2009
- Leblicq C. et al. Are Guidelines for Glucocorticoid Coverage in Adrenal Insufficiency Currently Followed? J Pediatr;158:492-8. 2011
- Malikova J. et al. Novel Insight into Etiology, Diagnosis and Management of Primary Adrenal Insufficiency. Horm Res Paediatr;
82:145–157. 2014
- Millar S. Clinical phenotypes of autoimmune polyendocrinopathy- candidiasis-ectodermal dystrophy seen in the Northern Ireland
paediatric population over the last 30 years. Ulster Med J; 81(3):118-122, 2012
- Perheentupa J. et al. Autoimmune Polyendocrinopathy-Candidiasis- Ectodermal Dystrophy. J Clin Endocrinol Metab 91: 2843–2850,
2006
- Perry R. et al. Primary Adrenal Insufficiency in Children: Twenty Years Experience at the Sainte-Justine Hospital, Montreal. J Clin
Endocrinol Metab 90: 3243–3250, 2005
- Polgreen LE. Early Diagnosis of Cerebral X-linked Adrenoleukodystrophy in Boys with Addison’s Disease Improves Survival and
Neurological Outcomes. Eur J Pediatr.170(8): 1049–105, 2011
- Roucher-Boulez F. et al. NNT mutations: a cause of primary adrenal insufficiency oxidative stress and extra- adrenal defects. EJE
175, 73–84, 2016.
- Simm PJ. et al. Primary adrenal insufficiency in childhood and adolescence: Advances in diagnosis and management. J. Paediatr.
Child Health 40, 596–599, 2004
- Tsai SL. et al. Primary Adrenocortical Insufficiency Case Series: Genetic Etiologies More Common than Expected. Horm Res Pae-
diatr; 85:35–42, 2016
7
3 - GENETIQUE DE L’INSUFFISANCE SURRENALE PRIMAIRE CONGENITALE
ROUCHER-BOULEZ Florence, MALLET-MOTAK Delphine, TARDY-GUIDOLLET Véronique, PLOT-
TON Ingrid, MOREL Yves.
UM Pathologies Endocriniennes Rénales Musculaires et Mucoviscidose – CPBE – Groupement Hospita-
lier Lyon-Est, 69677 LYON-BRON
L’insuffisance surrénale primaire (ISP) se caractérise par une atteinte de la corticosurrénale.
L’absence de gluco- et/ou minéralocorticoïdes expose au risque d’IS aiguë et menace le pronos-
tic vital. Elle diffère de l’IS secondaire, d’origine centrale. Chez l’adulte, elle est principalement
acquise et d’origine auto-immune. Chez l’enfant, 80% des formes sont d’origine génétique et se
révèlent le plus souvent durant la première année de vie. Dans ces formes, 90% des cas sont des
hyperplasies congénitales des surrénales (HCS) rapidement diagnostiquées à la naissance.
Parmi les 10% restants, sans surprise, les premiers gènes identifiés comme responsables sont
impliqués soit dans le développement de la surrénale (DAX-1/NR0B1, SF1/NR5A1), soit directe-
ment dans la biosynthèse des stéroïdes (STAR, CYP11A1, CYP21A2, HSD3B2, CYP11B2) ou la signali-
sation de l’ACTH (MC2R, MRAP). L’ISP peut être associée à une anomalie du développement sexuel
en fonction du niveau du blocage dans la stéroïdogenèse. L’avènement du séquençage massif en
parallèle (MPS) avec l’étude de l’exome (régions codantes du génome) a permis la découverte de
mutations dans des gènes non spécifiques de la surrénale, ouvrant le champ des recherches vers
de nouveaux mécanismes physiopathologiques.
En effet, des gènes découverts depuis 2012 : NNT, TXNRD2, PRDX3, GPX1, sont impliqués dans la
défense contre le stress oxydant. Le stress oxydant pouvant toucher de nombreux tissus, l’at-
teinte préférentielle de la surrénale chez l’homme reste inexpliquée mais impose un suivi rap-
proché des patients pour la prise en charge de possibles manifestations extra-surrénaliennes.
Pour preuve, ces découvertes ont permis de reconsidérer les mécanismes pathologiques dans
le syndrome triple A qui associe alacrymie, achalasie, IS et neurodégénérescence progressive,
suggérant une implication du stress oxydant. Nous rapportons dans notre cohorte de 18 patients
mutés NNT des cas d’hypothyroïdies, une cardiomyopathie hypertrophique, des anomalies go-
nadotropes et génitales. Un traitement par anti-oxydant, comme la N-acétylcystéine, pourrait
prévenir ou réduire de telles manifestations.
Le MPS a également permis d’identifier des gènes responsables d’ISP syndromiques : le gène
CDKN1C pour le syndrome IMAGe (Intrauterine growth restriction, Metaphyseal dysplasia, Adrenal
hypoplasia congenita and Genital anomalies), le gène SAMD9 pour le syndrome MIRAGE (Myelo-
dysplasia, Infection, Restriction of growth, Adrenal hypoplasia, Genital phenotypes, and Entero-
pathy). La transmission se fait respectivement par empreinte maternelle et de novo. Ces deux
syndromes sont dus à des mutations activatrices responsables d’une restriction de croissance par
inhibition du cycle cellulaire avec un défaut de la zonation surrénalienne. En revanche, les muta-
tions inactivatrices sont responsables d’un phénotype tout autre, sans atteinte surrénalienne. Le
gène MCM4, impliqué dans la réplication de l’ADN, a été mis en évidence dans une communauté
consanguine de gens du voyage irlandais associant l’ISP à une petite taille, des cassures chromo-
somiques anormalement fréquentes et un déficit en cellules immunitaires. Enfin, le dernier gène
découvert en 2017, SGPL1, est responsable lorsqu’il est muté d’une ISP associée à un syndrome
néphrotique et de possibles ichtyoses, hypothyroïdies, anomalies neurologiques. SGPL1 permet
la dégradation de la sphingosine 1-P (S1P) qui régule la migration, la différenciation et la survie
Les Insuffisances Surrénaliennes - 23 Juin 2017 8
cellulaire. D’autres mutations dans des protéines en amont de SGPL1 dans le métabolisme des
sphingolipides sont responsables de sphingolipidoses (Niemann Pick, Gaucher, Fabry) avec des
phénotypes rénaux décrits mais aucune atteinte surrénale.
Même si beaucoup de questions restent à élucider, ces découvertes génétiques sont fondamen-
tales car permettent une meilleure compréhension des mécanismes physiopathologiques im-
pliqués dans le maintien ou le développement de la surrénale. L’identification moléculaire est
importante pour confirmer le diagnostic étiologique et proposer un conseil génétique et une prise
en charge adaptées aux patients. Les données biologiques et cliniques sont indispensables pour
adapter la stratégie d’exploration des gènes. En dehors des HCS et de l’adrénoleucodystrophie
détectées par dosages biochimiques, on retrouve le plus souvent chez le garçon des mutations
DAX-1. Les autres pathologies de transmission autosomique récessive touchent par ordre de fré-
quence d’après notre cohorte, les gènes STAR, CYP11A1, NNT, MC2R, MRAP. Cependant des syn-
dromes peuvent être incomplets avec des chevauchements phénotypiques ne permettant pas
toujours l’analyse des gènes un par un, laissant alors une vraie place au MPS, panel de gènes voir
exome. Ils restent 5% des ISP congénitales à élucider.
Notes
94 - L’INSUFFISANCE SURRENALE AIGUE DE L’ENFANT
Claire Bouvattier, Endocrinologie pédiatrique, Centre de référence des maladies du développement
génital, Hôpital Bicêtre, Faculté de médecine Paris Sud.
L’insuffisance surrénale aiguë (ISA) est rare chez l’enfant. C’est une urgence diagnostique et thé-
rapeutique, qui met en jeu le pronostic vital. Le retard diagnostique est fréquent. Les signes cli-
niques d’ISA sont peu spécifiques : déshydratation extra cellulaire (pli cutané), hypotension arté-
rielle, tachycardie, marbrures, manifestations digestives (anorexie, douleurs abdominales parfois
pseudo-chirurgicales, nausées, vomissements, diarrhée) mais aussi fatigue et amaigrissement.
Les décompensations sont plus fréquentes chez les jeunes enfants, à l’occasion d’infections ba-
nales souvent digestives. Des hypoglycémies avec parfois des convulsions ne doivent pas égarer
le diagnostic chez les nourrissons et les petits enfants. Mélanodermie et taches ardoisées jugales
suggèrent une ISA périphérique chronique pre-existante. Le diagnostic doit être évoqué devant
une hyponatrémie à natriurèse conservée, une hyperkaliémie et une hypoglycémie. La mortalité
de l’ISA est mal connue mais non nulle chez l’enfant, mais l’on sait que le nombre d’hospitali-
sations pour ISA est plus fréquent dans l’IS primaire.. L’hyperplasie congénitale des surrénales
(HCS) correspond à environ la moitié des décompensations dues à une IS chronique primitive.
Dans l’HCS, sur une centaine d’enfants allemands de moins de 6 ans, un quart a été hospitalisé
une fois au cours de sa vie avec une hyponatrémie ou une hypoglycémie. La mortalité dans l’HCS,
évaluée en Suède, rapporte 13 décès d’ISA en pédiatrie entre 1952 et 2009. Avant traitement, et
sans retarder la prise en charge thérapeutique, ni attendre les résultats, sont réalisés des do-
sages de cortisol et ACTH. Une cortisolémie < 3 μg/dL (83 nmol/L) signe l’ISA. Le taux d’ACTH
précise le niveau d’atteinte de l’axe corticosurrénalien. La prise en charge est urgente, réalisée
au mieux en réanimation ou soins intensifs. Le traitement de l’ISA comprend une correction de
la déshydratation (G5 ou G10 si hypoglycémie, 10-15 meq/kg/j de sel, pas de potassium), le trai-
tement de l’hypovolémie par un remplissage si nécessaire, et l’administration d’hémisuccinate
d’hydrocortisone. Une surveillance étroite du patient est indispensable (scope, température, fré-
quence cardiaque, pression artérielle, diurèse, glycémie capillaire, ECG à la recherche de signes
d’hyperkaliémie, ionogrammes sanguins et urinaires pour quantification du bilan hydrosodé). Le
traitement préventif repose sur des recommandations simples et une éducation spécifique des
parents et/ou du patient selon son âge: port d’une carte d’insuffisant surrénalien, augmentation
de la supplémentation glucocorticoïdes en cas d’agression (fièvre, stress). L’éducation des pa-
rents du nourrisson est cruciale (administration du traitement, risque accru lors des épisodes
infectieux…). Les acquis et compétence doivent être évalués régulièrement.
Reisch N et al. Frequency and causes of adrenal crises over lifetime in patients with 21-hydroxylase deficiency. Eur J Endocrinol 2012,
167: 35-42.
Hsieh S et al. Presentation of primary adrenal insufficiency in childhood. J Clin Endocrinol Metab 2011, 96: 1945-48.
Odenwald B et al. Children with classic congenital adrenal hyperplasia experience salt loss and hypoglycemia: evaluation of adrenal
crises during the first 6 years of life. Eur J Endocrinol 2016, 174: 177-186.
Les Insuffisances Surrénaliennes - 23 Juin 2017 10Notes
115 - IMPACT D’UNE TRANSITION REUSSIE SUR LA QUALITE DE VIE DES PATIENTS AVEC HCS
Pr Philippe Touraine
Chef du Service d’Endocrinologie et Médecine de la Reproduction
Equipe du Centre de Référence des Maladies Endocriniennes Rares de la Croissance
Equipe du Centre de Référence des Maladies Rares Gynécologiques
Equipe Européenne ENDO-ERN
Plusieurs études se sont déjà attardées sur le retentissement de l’hyperplasie congénitale des
surrénales sur la qualité de vie ; toutefois aucune étude n’a été menée à ce jour chez les patients
avec HCS pendant la période de transition. Nous avons eu l’occasion de mener une étude chez
nos patients présentant une HCS classique ou non classique révélée pendant l’enfance, nés entre
1970 et 1990 et qui avaient été initialement suivis par des services de pédiatrie de la région Ile de
France. Alors que nous avions une population estimée à 183 patients incluables dans l’étude,
seuls 73 d’entre eux ont pu répondre aux questionnaires de qualité de vie ce qui souligne bien
d’emblée la difficulté de suivre cette cohorte de patients. Au sein de cette population, 81% avaient
été adressés directement par un service d’endocrinologie pédiatrique. Les réponses aux ques-
tionnaires de qualité de vie pouvaient apparaître similaires si on comparait les patients ayant
effectué une transition et ceux qui ne l’avaient pas fait. Mais si au-delà du transfert d’un service
pédiatrique vers un service adulte, si on s’intéresse aux patients qui ont un suivi régulier, il est
frappant de noter que ces derniers ont une meilleure santé physique, psychologique, un score
d’intégration environnementale et une qualité globale de vie supérieures à ceux qui n’ont pas de
suivi régulier. Ces données méritent d’être confirmées mais ne font que souligner l’importance
d’un suivi régulier de ces patients avec maladies rares dès lors que celui-ci peut être associé à
une meilleure qualité de vie.
Notes
Les Insuffisances Surrénaliennes - 23 Juin 2017 12Notes
136 - RELATIVE ADRENAL INSUFFICIENCY IN THE ICU: FACT OR FICTION ?
Greet Van den Berghe MD, PhD -- Department of Intensive Care Medicine, KU Leuven,
B-3000 Leuven, BELGIUM
Critical illness is hallmarked by hypercortisolemia, traditionally attributed to stress-induced HPA
axis activation. “Relative” failure of this activation, suggested to be identifiable by a reduced cor-
tisol response to ACTH injection, is presumed to require treatment. However, as plasma ACTH
concentrations are mostly low during critical illness rather than high, we hypothesized that re-
duced cortisol metabolism could play a major role, which could reshape the understanding of this
so-called “relative” adrenal failure. We found that cortisol production rates were hardly elevated
if at all during critical illness. In contrast, cortisol clearance was found uniformly reduced to less
than half the values in healthy subjects, determining the 3.5-fold increased plasma cortisol. Re-
duced cortisol metabolism was due to reduced breakdown of cortisol by 5β- and 5α-reductases
in liver and by 11β-HSD2 in kidney. This, in turn, was explained by elevated bile-acids, uniformly
present during critical illness in the face of suppressed expression of the nuclear receptors known
to function as bile-acid sensors. With time, the low plasma ACTH may cause adrenal atrophy, as
suggested by a human postmortem study revealing signs of ACTH deficiency and loss of adreno-
cortical integrity and function in prolonged, not acute, critically ill patients.
In conclusion, during critical illness, elevated bile-acids may reduce cortisol breakdown which
drives hypercortisolemia and leads to ACTH suppression by feedback inhibition. These findings
have several important clinical implications for the management of ICU patients. The role of bile-
acids and its regulation during critical illness should be further explored.
Notes
Les Insuffisances Surrénaliennes - 23 Juin 2017 14Notes
157 - LES « TAKE HOME MESSAGES » DU CONSENSUS FRANÇAIS POUR LA PRISE EN CHARGE
DE L’INSUFFISANCE SURRÉNALE.
Yves Reznik, Service d’endocrinologie CHU Caen
Centre de compétence des maladies rares de la surrénale
La SFE et la SFEDP ont élaboré des recommandations sur la prise en charge de l’insuffisance
surrénale qui ont été communiquées au congrès de la SFE en octobre 2015 et qui seront publiées
in extenso en 2017. Ces recommandations se sont basées sur une analyse de la littérature et
sur les opinions de 18 experts, 11 endocrinologues adultes, 6 endocrinologues pédiatres et un
interniste, qui ont travaillé en sous-groupes sur 6 thématiques comprenant l’épidémiologie, le
diagnostic positif, le diagnostic étiologique, l’insuffisance surrénale aigue, le traitement, l’éduca-
tion thérapeutique et le suivi. Par comparaison aux consensus existants nord-américain et eu-
ropéen qui se sont limités au champ de l’insuffisance surrénale primaire de l’adulte, Le champ
des recommandations a été élargi à l’insuffisance surrénale primaire et secondaire, de l’enfant
et de l’adulte. Pour chacune des recommandations, le groupe s’est attaché à définir la force de la
recommandation et son niveau de preuve. La présentation s’attachera à rapporter les principales
recommandations, notamment celles concernant les stratégies du diagnostic positif et étiolo-
gique, les valeurs seuils des tests diagnostics, les modalités du traitement aigu et chronique, en-
fin les bases de l’éducation thérapeutique, de la prévention de la crise aigue et de la surveillance
au long cours du patient souffrant d’insuffisance surrénale lente.
Notes
Les Insuffisances Surrénaliennes - 23 Juin 2017 16Notes
178 - CAS CLINIQUE D’INSUFFISANCE SURRENALE CHEZ L’ADULTE
Delphine Vezzosi, service d’endocrinologie CHU Toulouse
Centre de compétence des maladies rares de la surrénale
Le traitement de l’insuffisance surrénalienne périphérique chronique repose sur l’association
d’un traitement par glucocorticoïdes et d’un traitement par minéralocorticoïdes. On oublie par-
fois le troisième pilier d’un traitement substitutif bien conduit : une alimentation normo-salée.
M G. est un patient de 57 ans présentant un cancer du rein multimétastatique pour lequel il a
bénéficié en 2004 et 2007 d’une surrénalectomie bilatérale en deux temps pour des métastases
surrénaliennes. Un traitement par Hydrocortisone 10 mg matin et 10 mg midi et Fludrocortisone
50 μg/jour a été institué au décours de la deuxième surrénalectomie. L’observance du traitement
a toujours été excellente. Il n’y a jamais eu de décompensation surrénalienne aigue. Il a ensuite
bénéficié, sur le plan tumoral, en 2010 d’une radiothérapie oculaire pour des métastases ocu-
laires, d’un enclouage centro-médullaire de l’humérus gauche en 2011 et du fémur gauche en
2013 pour des fractures pathologiques. Un traitement systémique à visée anti-tumorale à base
de bevacizumab (’Avastin®) a été réalisé pendant 6 mois. Il a enfin bénéficié d’une radiothérapie
osseuse à visée antalgique pendant 15 jours. Le traitement par hydrocortisone a été remplacé
juste avant la radiothérapie par du Solupred 100 mg/24h avec décroissance progressive jusqu’à
20 mg/24h depuis 7 jours. Cette radiothérapie a entrainé des troubles importants de l’appétit,
sans vomissements et sans diarrhée. Il existe en revanche une anorexie complète et M G. ne se
nourrit depuis une semaine que d’un complément alimentaire par 3 Delical® de 200 ml par jour.
L’observance du traitement substitutif reste bonne.
Il est adressé aux urgences pour une altération de l’état général associé à des douleurs abdomi-
nales diffuses. La tension artérielle à l’entrée est à 100/60 mmHg. La natrémie est à 130 mmol/l
avec une kaliémie à 5.3 mmol/l. Le traitement qu’il poursuit à l’entrée comporte du Solupred 20
mg/jour associé à de la Fludrocortisone 50 μg/jour.
Une réhydratation intra-veineuse par sérum salé isotonique et substitution par hydrocortisone à
100 mg/24h en continu est débuté permettant une résolution rapide de la symptomatologie ainsi
qu’une normalisation rapide des perturbations hydroélectrolytiques. Le diagnostic de décompen-
sation surrénalienne aigue est retenu. Aucun facteur déclenchant infectieux n’est retrouvé.
En revanche l’apport alimentaire exclusif par les 600 ml quotidien de Delical®, responsable d’une
alimentation quasi désodée (0.3 g de sel/jour) semble être le facteur déclenchant le plus pro-
bable. Un régime désodé peut en effet être la cause exclusive d’une décompensation surréna-
lienne aigue chez un patient insuffisant surrénalien chronique. Cela est parallèlement l’occasion
de rappeler que l’activité minéralocorticoïde des substitutions glucocorticoïdes varie en fonction
du traitement utilisé. Ainsi, à dose équivalente, alors que le traitement par hydrocortisone a une
activité minéralocorticoide relative de 1, la prednisolone a une activité minéralocorticoide relative
comprise entre 0,6 et 0,8 imposant, le plus souvent, une
augmentation de la posologie de fludrocortisone ou au moins une réévaluation de la substitution
minéralocorticoide lors du remplacement de l’hydrocortisone par les glucocorticoïdes de syn-
thèse [1]. Le traitement par Fludrocortisone à 50 μg/jour que M G. poursuivait était donc probable-
ment adapté lors de la substitution par Hydrocortisone mais était vraisemblablement insuffisant
lors de la substitution par la prednisolone. Cette substitution insuffisante par minéralocorticoïdes
a été responsable d’une situation d’hypovolémie chronique, très fréquemment rapporté chez les
Les Insuffisances Surrénaliennes - 23 Juin 2017 18patients insuffisants surrénaliens chroniques substitués par de la prednisolone [2, 3]. Cette hypo-
volémie chronique est un facteur favorisant majeur des décompensations surrénaliennes aigues.
Une aggravation de l’hypovolémie par une restriction sodée surajoutée entraine alors très rapide-
ment la décompensation surrénalienne aigue [4].
En conclusion, cette observation est l’occasion de rappeler la nécessité d’un régime normosodé
chez tous les patients insuffisants surrénaliens chroniques. Une éducation soigneuse du patient
et de son entourage concernant ce point est une étape indispensable à la bonne prise en charge
de l’insuffisance surrénalienne chronique. Cette observation est également l’occasion de rappeler
la nécessité d’une substitution minéralocorticoide suffisante permettant d’éviter l’hypovolémie
chronique, facteur favorisant des décompensations aigues.
Références
[1]. Grossmann C, Scholz T, Rochel M, Bumke-Vogt C, Oelkers W, Pfeiffer AF, Diederich S, Bahr V. Transactivation via the human
glucocorticoid and mineralocorticoid receptor by therapeutically used steroids in CV-1 cells: a comparison of their glucocorticoid
and mineralocorticoid properties. Eur J Endocrinol 2004; 151:397-406
[2]. Jadoul M, Ferrant A, De Plaen JF, Crabbe J. Mineralocorticoids in the management of primary adrenocortical insufficiency. J
Endocrinol Invest 1991; 14:87-91
[3]. Smith SJ, Macgregor GA, Markandu ND, Bayliss J, Banks RA, Prentice MG, Dorrington-Ward P, Wise P. Evidence that patients with
Addison’s disease are undertreated with fludrocortisone. Lancet 1984; 1:11-14
[4]. Hahner S, Loeffler M, Bleicken B, Drechsler C, Milovanovic D, Fassnacht M, Ventz M, Quinkler M, Allolio B. Epidemiology of adrenal
crisis in chronic adrenal insufficiency: the need for new prevention strategies. Eur J Endocrinol 2010; 162:597-602
Notes
199 - MORBIDITE DE L’INSUFFISANCE SURRENALE CHEZ LES PATIENTS HOSPITALISES
Gudmundur Johannsson, MD, Ph.D
Professor of Endocrinology, Institute of Medicine, Sahlgrenska Academy, University of Gothenburg, and
Sahlgrenska University Hospital, Gothenburg, Sweden
Before the availability of hydrocortisone, the majority of patients with primary adrenal insuffi-
ciency (AI) died within 2 years of diagnosis. However, after the discovery hydrocortisone life-long
replacement therapy could be obtained with the assumption that life expectancy would be normal.
More recent Scandinavian studies have, however, shown a 2-fold increase in standardized morta-
lity in patients with Addison’s disease [1]. Moreover, patients with hypopituitarism and secondary
adrenal insufficiency due to various causes [2], treated acromegaly [3] and patients with long-term
cure of Cushing’s disease [4] have all markedly increased mortality in comparison with those not
in need of glucocorticoid replacement. The increased mortality observed in both primary and
secondary AI is largely due to cardiovascular diseases and infections.
Most previous studies on morbidity in AI is based on European cohorts showing increased hy-
pertension, metabolic syndrome, reduced quality of life and increased hospital admission rates
due to infections and high rate of adrenal crisis [1]. A recent study using a US-based national payer
database identified 1,014 with primary AI and 8,818 with secondary AI [5]. The study showed that
patients with AI had higher odds of diabetes mellitus, hypertension, hyperlipidaemia, depression
and anxiety than matched controls. Inpatient admissions were also 4-times more frequent in
patients with AI than controls with infection being the dominant cause for admission. These large
US-based data support previous European data showing and increased incidence of infectious
episodes among patients with primary AI with an adjusted incidence risk ratio of 1.58 (95% CI
1.47–1.70). Also, the adjusted incidence risk ratio for hospitalisation due to an infectious episode
was 4.34 (95% CI 3.04–6.22) showing similar to the US-based data that the risk is at least 4-fold
among patients with AI to be hospitalised due to an infection.
Most patients with hypopituitarism and AI also have other hormone deficiencies that may in-
fluence outcome, but also patients with autoimmune primary AI have an increased burden of
other autoimmune diseases [6]. Approximately half of patients with primary AI also have another
autoimmune disorder, mostly autoimmune thyroid disease that does not seem to have a signifi-
cant impact on mortality [7]. Having both primary AI and type 1 diabetes may occur in approxima-
tely 10% of patients with AI. This rare combination has recently been shown to be associated with
a more than 4-times excess mortality than having diabetes alone again mainly associated with
cardiovascular, infectious and sudden deaths [8].
There is increasing evidence that patients with AI have an increased burden of disease and excess
mortality that is mainly due to infections and cardiovascular disease. One patient population in
particular seem to have a marked increased mortality rate and that are patients with the rare
combination of primary AI and diabetes. These data clearly demonstrate that there is a need for
improvement in the management of these patients in particular in relation to their daily mainte-
nance dose of glucocorticoid as well as in the intercurrent illness management.
References
[1] J ohannsson G, Falorni A, Skrtic S, et al. Adrenal insufficiency: review of clinical outcomes with current glucocorticoid replacement
therapy. Clin Endocrinol (Oxf) 2015; 82:2-11
[2] G aillard RC, Mattsson AF, Akerblad AC,et al. Overall and cause-specific mortality in GH-deficient adults on GH replacement. Eur
J Endocrinol 2012; 166:1069-1077
Les Insuffisances Surrénaliennes - 23 Juin 2017 20[3] S herlock M, Reulen RC, Alonso AA, et al. ACTH deficiency, higher doses of hydrocortisone replacement, and radiotherapy are
independent predictors of mortality in patients with acromegaly. J Clin Endocrinol Metab 2009; 94:4216-4223
[4] C layton RN, Jones PW, Reulen RC, et al. Mortality in patients with Cushing’s disease more than 10 years after remission: a multi-
centre, multinational, retrospective cohort study. Lancet Diabetes Endocrinol 2016; 4:569-576
[5] S tewart PM, Biller BM, Marelli C, et al. Exploring Inpatient Hospitalizations and Morbidity in Patients With Adrenal Insufficiency. J
Clin Endocrinol Metab 2016; 101:4843-4850
[6] D alin F, Nordling Eriksson G, et al. Clinical and Immunological Characteristics of Autoimmune Addison Disease: A Nationwide
Swedish Multicenter Study. J Clin Endocrinol Metab 2017; 102:379-389
[7] B ensing S, Brandt L, Tabaroj F, et al. Increased death risk and altered cancer incidence pattern in patients with isolated or com-
bined autoimmune primary adrenocortical insufficiency. Clin Endocrinol (Oxf) 2008; 69:697-704
[8] C hantzichristos D, Persson A, Eliasson B, et al. Mortality in patients with diabetes mellitus and Addison’s disease: a nationwide,
matched, observational cohort study. Eur J Endocrinol 2017; 176:31-39
Notes
2110 - INSUFFISANCE SURRENALE ET GROSSESSE
Frédéric CASTINETTI - Hôpital de la Conception, Marseille, France
Centre de référence des maladies rares de l’hypophyse (HYPO)
La grossesse est une situation particulière pour la patiente porteuse d’une insuffisance surréna-
lienne, mais également pour son endocrinologue, qui peut se poser des questions sur la prise en
charge optimale substitutive au vu de la rareté de cet évènement. Après avoir abordé l’adaptation
physiologique surrénalienne au cours de la grossesse, nous discuterons des conséquences qui
en découlent en cas d’insuffisance surrénalienne, avec en particulier la question de l’adaptation
des doses d’hydrocortisone en cours et en fin de grossesse, et de la prise en charge de l’accou-
chement et de l’allaitement. Nous aborderons la question de la difficile surveillance des signes de
sous-dosage ou surdosage pendant la grossesse.
Notes
Les Insuffisances Surrénaliennes - 23 Juin 2017 22Notes
2311 - CONTINUOUS SUBCUTANEOUS HYDROCORTISONE INFUSION BY PUMP OR MODIFIED
RELEASE HYDROCORTISONE: WHAT IS THE BEST FOR TREATING ADDISON’S DISEASE?
Marianne Øksnes, MD, phD. Haukeland University hospital, University of Bergen, Norway
When Thomas Addison in the 19th century first described the clinical picture of Addison’s disease
(AD) no treatment was available and the disease was always lethal [1]. Although efforts were made
to produce adrenal extracts to prolong survival, the real treatment revolution came with the identi-
fication of the different adrenal steroids by Reichstein, Hench and Kendall, for which they won the
Nobel Prize Award in 1950. Since then, the replacement therapy remained almost unchanged for
decades. Oral hydrocortisone (OHC) is the most common treatment choice worldwide [2]. Corti-
sone acetate (CA) is considered equally effective as OHC and is preferred in countries where OHC
is not easily commercially available. In some countries, long-acting GCs such as prednisolone
and dexamethasone are also used, despite the unphysiological pharmacodynamic effects of these
drugs.
Over the last decades, systematic cohort and registry studies have revealed reduced HRQoL, an
unfavourable metabolic profile and increased mortality in patients with AD [3). Currently, unphy-
siological or insufficient GC replacement is believed to be a major explanation [4, 5], but other
causes may be the autoimmune disease per se, co-existing hormone deficiencies or unknown
factors [6-8]. These concerns have sparked interest in improving the therapeutic regimes and
have resulted in several new treatment options.
Plenadren®, recently licensed in Europe, is a modified release formulation of hydrocortisone that
provides the potential for once daily dosing. It has an immediate release coating combined with an
extended-release core, which provides once daily morning dosing. The bioavailability of Plenad-
ren® is 20% less than oral hydrocortisone and dose adjustment may be required. A recent study
demonstrated that 24 h exposure of cortisol was reduced and associated with reduction of body
weight, blood pressure and HbA1c [9]. Also, improvement in HRQoL was noted. Although such
treatment successfully restored daytime cortisol levels to normal, the physiological late night in-
crease in cortisol is not re-established.
Chronocort® is a modified-release hydrocortisone preparation that replaces the overnight rise
in cortisol levels [10, 11]. A recent phase 2 study in CAH patients showed that a twice daily “too-
thbrush” regimen (10 mg morning and 20 mg evening) resulted in a daily cortisol AUC similar to
healthy individuals and a delayed Cmax cortisol level mimicked the normal physiological morning
cortisol peak [11]. Potential effects on health and well-being in AD will have to be investigated in
future studies.
Analogous to insulin pump treatment in diabetes, continuous subcutaneous hydrocortisone infu-
sion (CSHI) has been introduced as a treatment option for AD and CAH [12, 13]. In a controlled trial
in 34 patients, we found that CSHI was safe, allowed restoration of the circadian biorhythm and
improved HRQoL in AD patients, although the last finding was not reproduced in a smaller blinded
trial [14]. Today, CSHI is a treatment option for selected AD patients, when other treatments have
failed. With improvement of pump technology, and reduced costs, CSHI may become an affordable
choice for a larger group in the future.
Les Insuffisances Surrénaliennes - 23 Juin 2017 24[1] A ddison T. On the constitutional and local effects of disease of the supra-renal capsules. Birmingham, Alabama, USA: Leslie B.
Adams Jr., Division of Gryphon Editions, Ltd.; 1855.
[2] D ebono M, Ross RJ. Doses and steroids to be used in primary and central hypoadrenalism. Ann Endocrinol (Paris). 2007;68(4):265-
7.
[3] H usebye ES, Allolio B, Arlt W, Badenhoop K, Bensing S, Betterle C, et al. Consensus statement on the diagnosis, treatment and
follow-up of patients with primary adrenal insufficiency. J Intern Med. 2013.
[4] G rossman A, Johannsson G, Quinkler M, Zelissen P. Perspectives on the management of adrenal insufficiency: Clinical insights
from across Europe. Eur J Endocrinol. 2013.
[5] H ahner S, Loeffler M, Fassnacht M, Weismann D, Koschker AC, Quinkler M, et al. Impaired subjective health status in 256 patients
with adrenal insufficiency on standard therapy based on cross-sectional analysis. J Clin Endocrinol Metab. 2007;92(10):3912-22.
[6] B ianchi GP, Zaccheroni V, Solaroli E, Vescini F, Cerutti R, Zoli M, et al. Health-related quality of life in patients with thyroid disor-
ders. Qual Life Res. 2004;13(1):45-54.
[7] Kvien TK, Kaasa S, Smedstad LM. Performance of the Norwegian SF-36 Health Survey in patients with rheumatoid arthritis. II. A
comparison of the SF-36 with disease-specific measures. J Clin Epidemiol. 1998;51(11):1077-86.
[8] N icolucci A, Maione A, Franciosi M, Amoretti R, Busetto E, Capani F, et al. Quality of life and treatment satisfaction in adults with
Type 1 diabetes: a comparison between continuous subcutaneous insulin infusion and multiple daily injections. Diabetic medicine :
a journal of the British Diabetic Association. 2008;25(2):213-20.
[9] J ohannsson G, Nilsson AG, Bergthorsdottir R, Burman P, Dahlqvist P, Ekman B, et al. Improved cortisol exposure-time profile and
outcome in patients with adrenal insufficiency: a prospective randomized trial of a novel hydrocortisone dual-release formulation.
J Clin Endocrinol Metab. 2012;97(2):473-81.
[10] V erma S, Vanryzin C, Sinaii N, Kim MS, Nieman LK, Ravindran S, et al. A pharmacokinetic and pharmacodynamic study of de-
layed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital
adrenal hyperplasia. Clin Endocrinol (Oxf). 2010;72(4):441-7.
[11] M allappa A, Sinaii N, Kumar P, Whitaker MJ, Daley LA, Digweed D, et al. A phase 2 study of Chronocort, a modified-release
formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab.
2015;100(3):1137-45.
[12] O ksnes M, Bjornsdottir S, Isaksson M, Methlie P, Carlsen S, Nilsen RM, et al. Continuous subcutaneous hydrocortisone infusion
versus oral hydrocortisone replacement for treatment of addison’s disease: a randomized clinical trial. J Clin Endocrinol Metab.
2014;99(5):1665-74.
[13] N ella AA, Mallappa A, Perritt AF, Gounden V, Kumar P, Sinaii N, et al. A Phase 2 Study of Continuous Subcutaneous Hydrocorti-
sone Infusion in Adults With Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab. 2016;101(12):4690-8.
[14] G agliardi L, Nenke MA, Thynne TR, von der Borch J, Rankin WA, Henley DE, et al. Continuous subcutaneous hydrocortisone in-
fusion therapy in Addison’s disease: a randomized, placebo-controlled clinical trial. J Clin Endocrinol Metab. 2014;99(11):4149-57.
Notes
2512 - TOLLS FOR THERAPEUTIC PATIENT EDUCATION IN ADRENAL INSUFFICIENCY.
Dr Laurence Guignat, Service des Maladies Endocriniennes et Métaboliques, Centre de Référence des
Maladies Rares de la Surrénale, Hôpital Cochin, 27, rue du Faubourg St-Jacques, 75014 Paris, France.
Email : laurence.guignat@cch.aphp.fr
Therapeutic patient education programs have been developed these last years by several teams
for patients with adrenal insufficiency. Tools have also been developed by expert physicians and
the association of patients with the help of educational professionals. The pedagogical tools cor-
respond to the educational material useful to animate the workshops. A focus will be given to pro-
gram tools to avoid or treat early acute adrenal insufficiency, with some examples, without being
exhaustive. These tools have been developed to achieve the following objectives: 1) Carry on the
security tools; 2) Identify risk situations, signs of acute adrenal insufficiency; 3) Know how to adapt
oral glucocorticoid treatment; 4) Know how to make a hydrocortisone subcutaneous injection; 5)
Know how to adapt treatment in special situations: heat, physical exercise, travel, ... 6) Use the
resources of the health care system in a relevant way.
Worshops Main objectives Tools
Better know your illness - List signs of acute adrenal insuffi- Post’it meeting (Métaplan®)
ciency Photovoice
- Recognize your own early signs of The Narrative of acute adrenal insuffi-
acute adrenal insufficiency ciency by (one) patient(s)
- Explain the differences between hy- Schematic representation of equiva-
drocortisone and anti-inflammatory lences
corticosteroids Playing cards info/intox
Manage your treatment -Recognize situations at risk that re- Playing cards risk situations
quire increased treatment Quizz
-Know how to adapt oral glucocorticoid Timeline
treatment
- Know when and how to inject hydro- Preparation and injection training;
cortisone notice / video
-Know how to make your “emergency Tools
kit”
Live with your illness - Cope with difficult situations Round of Decisions
- Adapt your daily life Role games
- Identify resources List of resources
Individualized school project
Les Insuffisances Surrénaliennes - 23 Juin 2017 26Many tools are now available to teams. However, the implementation of therapeutic patient edu-
cation is hampered by many practical problems: the lack of time and human resources to train
medical and paramedical staff in the animation of workshops and the use of tools, the lack of
support in the authorization procedure, the unregulated modes of funding and organisation.
Notes
2713 - L’INSUFFISANCE SURRENALE AIGUE VUE PAR LES PATIENTS : RESULTATS D’UNE
ENQUETE SUR L’ISA AUPRES DES ADHERENTS DE L’ASSOCIATION SURRENALES.
Claudine Colin, Christine Rougeau
L’association Surrénales, créée le 12 septembre 1996 et déclarée d’intérêt général le 11 mai 2006
compte plus de 730 adhérents.
L’association est organisée en 5 entités : HCS et hypoplasie congénitale des surrénales, maladie
d’Addison, insuffisance surrénale secondaire, Cushing et corticosurrénalome, phéochromocy¬-
tome et hyperaldostéronisme.
Ses objectifs sont d’informer les malades avec des documents, plaquettes, livrets d’information
rédigés en collaboration avec des experts des Centres de Référence / Compétence.
Elle œuvre pour l’amélioration de la qualité de vie des malades.
Elle organise régulièrement des sondages auprès de ses adhérents pour recueillir les besoins
prioritaires des patients.
Suite au retour depuis quelques années d’un certain nombre d’adhérents de l’association sur
une prise en charge inadaptée de la crise d’insuffisance surrénale aigue (ISA), l’association Sur-
rénales a élaboré et diffusé un questionnaire auprès de ses 730 adhérents au mois de mars 2017 :
410 adhérents ont répondu à ce questionnaire sur l’ISA soit un taux de participation de 56,16% !
Ce sujet emporte un vif intérêt des adhérents de l’association Surrénales en raison du caractère
vital et donc important qu’il revêt.
Le questionnaire a été organisé autour de 4 thèmes:
1. Participation aux ateliers d’éducation thérapeutique pour apprendre la gestion de l’ISA.
2. Connaissance des modalités d’injection d’hydrocortisone en urgence.
3. Retours d’expériences sur le système actuel d’injection d’urgence et la gestion de la situa-
tion d’urgence.
4. Améliorations souhaitées à la prise en charge de l’ISA.
Conclusion: les patients atteints d’insuffisance surrénale espèrent vivement que les résultats de
cette enquête apporteront des pistes pour une prise en charge de l’insuffisance surrénale aigue
plus rapide et en adéquation avec le consensus de 2015
Les Insuffisances Surrénaliennes - 23 Juin 2017 28Notes
29Notes Les Insuffisances Surrénaliennes - 23 Juin 2017 30
Notes
31Notes Les Insuffisances Surrénaliennes - 23 Juin 2017 32
www.surrenales.com
Journée de l’Association Surrénales
Les Insuffisances
Surrénaliennes
Avec le parrainage de la SFE et de la SFEDP
Amphithéâtre Pierre Laroque
Ministère des Solidarités et de la Santé
14, avenue Duquesne - Paris 7e
Création et mise en page : La Com et Plus - 06 08 32 78 18 - ©Paris 2017 - 06
En partenariat avec les Centres de Référence
labellisés pour ces maladies et la filière Firendo.
avec le soutien institutionnel des Laboratoires
H.A.C. Pharma
we develop the futureVous pouvez aussi lire

















































