ASSEMBLAGE ET ANNOTATION DE GÉNOMES EUCARYOTES - 7 SEPTEMBRE 2015
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous

ASSEMBLAGE ET ANNOTATION DE GÉNOMES EUCARYOTES M2 BC2T | Benjamin Noel (bnoel@genoscope.cns.fr) 7 SEPTEMBRE 2015

WORKFLOW DES PROJETS DE SÉQUENÇAGE
NGS-RG
Laboratory
Information
Management
*.fastq.gz raw System
Backup
NGS-QC
*.fastq.gz clean
Genome computing
assembly facilities
Gene
prediction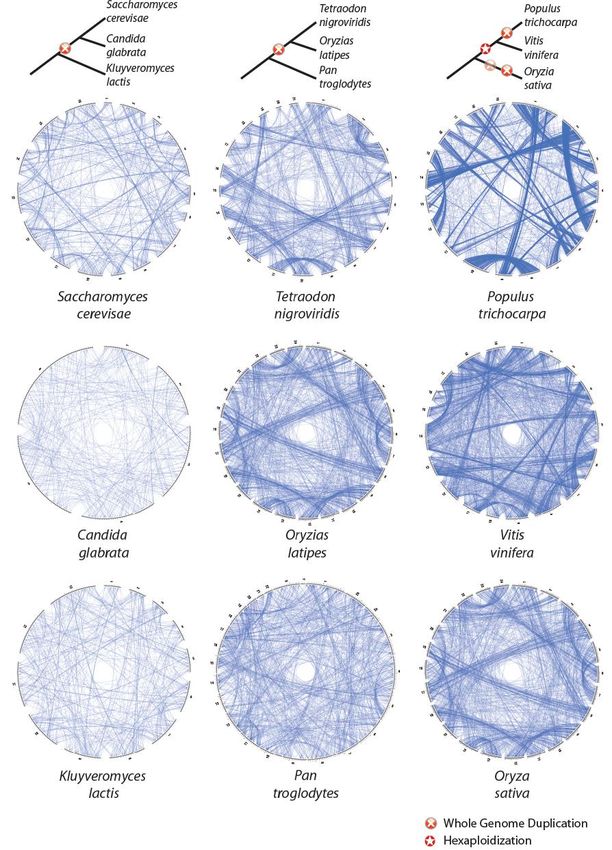
ASSEMBLAGE ET ANNOTATION DE GÉNOMES
EUCARYOTES
1. Introduction à l’assemblage de génomes
2. Annoter un génome Eucaryote : théorie et réalité
3. L'annotation automatique au Genoscope
4. Projets de génomique Eucaryotes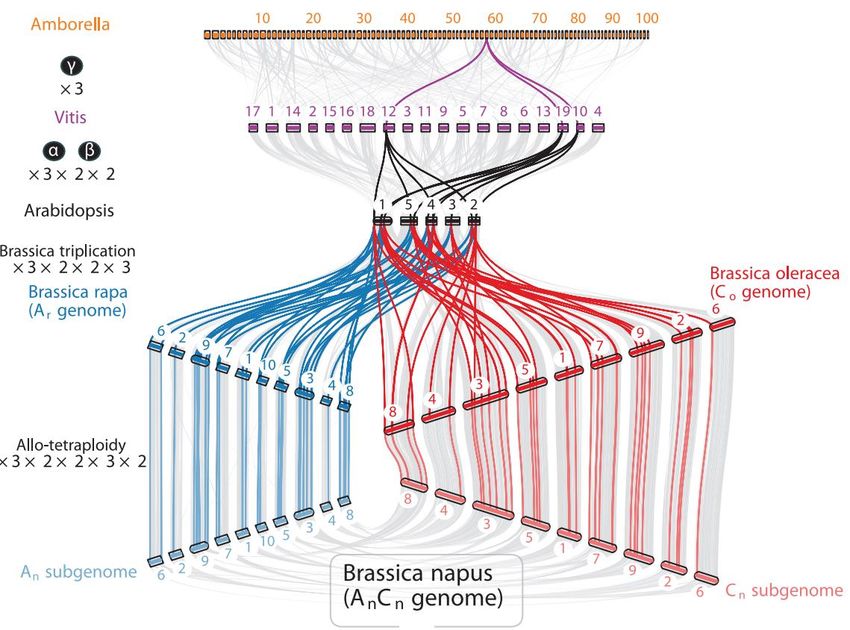
INTRODUCTION À L’ASSEMBLAGE
Building a genome
Genome DNA abundance Type of library Sequencing Assembly Assembly QC
complexity and quality technology method
Ploïdy Material unlimited Overlapping Sanger de bruijn Graph Continuity
reads (DGB)
Zygoty Very small 454 Missamblies
organisms Single reads Overlap-Layout-
Repeats Illumina Consensus Physical map
Monoclonal Short fragments (OLC)
Genome size population Ion Torrent Genetic map
Long fragments Scaffolding
High molecular PacBio
weight DNA BAC Gapclosing
Nanopore
Nuclear DNA
contamination
INTRODUCTION À L’ASSEMBLAGE
Complexité d’un génome
1. A quoi ressemble le génome:
« Le génome à une taille d’environ 500 Mb, est diploïde, à 99 % homozygote et
contient environ 40% de séquences répétées »
2. Définir les objectifs qualités de l’assemblage final:
Au moins 95% des gènes sont dans l’assemblage du génome
Au moins 90 % du génome est contenu dans l’assemblage
N50 contig > taille moyenne d’un gène
N50 scaffold > 1 Mb
Moins de 5% de bases indéterminées
3. Définition d’une stratégie de séquençage et d’assemblage
Accès ADN haut poids moléculaire ? => long reads, long fragments, optical maps
« easy » genome : Illumina sequencing seulement
« hard » genome : stratégie hybride
Quelles technologies de séquençage sont disponibles ? Quels sont leurs coûts ?
Quels compromis faire ?
INTRODUCTION À L’ASSEMBLAGE Assemblage de génome Définition: Obtenir les séquences qui sont le plus proches possibles de celles des chromosomes.
INTRODUCTION À L’ASSEMBLAGE
AAAAAA
Assemblage de génome : Répétitions AAAAAA
AAAAAA
AAAAAAAAAAAAAAAAAAAAAAA AAAAAA
AAAAAA AAAAAA AAAAAA AAAAAA
reads AAAAAA contigs
AAAAAA AAAAAA AAAAAA
AAAAAA AAAAAA Assemblage AAAAAA
AAAAAA
AAAAAA
Repeat Repeat
contig 1 contig 2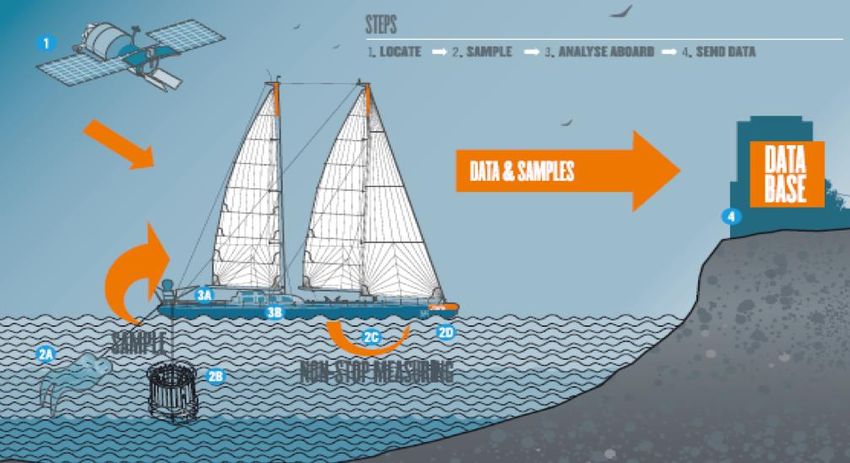
INTRODUCTION À L’ASSEMBLAGE
Assemblage de génome : Hétérozygotie
Chromosome 1 allèle A
Chromosome 1 allèle B
Assemblage
INTRODUCTION À L’ASSEMBLAGE
Overlap Layout Consensus (OLC)
Genome Reads
GCGTATAAGCTATACGCT GCGTATA
TATAAGCTATA
TATACG
TACGCT
Layout
Overlap
GCGTATA TATAAGCTATA
GCGTATA TATAAGCTATA
TATAAGCTATA TATACG
TATACG
GCGTATA TATACG
TACGCT TATACG TACGCT
Consensus
GCG TATA AGC CGCT
INTRODUCTION À L’ASSEMBLAGE
De Bruijn Graph (DBG)
Genome Reads GCGT
CGTA
GTAT
4-mers
GCGTATAAGCTATACGCT GCGTATA TATA
ATAA
TATAAGCTATA TAAG
AAGC
TATACG AGCT
GCTA
TACGCT CTAT
ATAC
TACG
ACGC
CGCT
4-mers overlap graph
Chaque k-mer est un nœud. 2 nœud sont connectés si les k-mers se chevauchent par k-1 mer
ATAA TAAG
GCGT CGTA GTAT
AAGC
TATA
AGCT
CTAT GCTA
CGCT ACGC TACG ATACINTRODUCTION À L’ASSEMBLAGE De Bruijn Graph (DBG)
INTRODUCTION À L’ASSEMBLAGE
OLC vs DBG
DBG
Taille de k-mer limite
Utile pour les reads < 200 bp
Casse les contigs dans les régions répétées et/ou hétérozygotes
Algorithme rapide mais gourmand en mémoire
OLC
Taille reads > 200 bp
Casse les contigs dans les régions répétées et/ou hétérozygotes
Algorithme lent mais faible consommateur en mémoireASSEMBLAGE ET ANNOTATION DE GÉNOMES
EUCARYOTES
1. Introduction à l’assemblage de génomes
2. Annoter un génome Eucaryote : théorie et réalité
3. L'annotation automatique au Genoscope
4. Projets de génomique EucaryotesANNOTER UN GÉNOME EUCARYOTE: THÉORIE
Annoter une séquence, c’est donner un sens biologique à celle-ci
Plusieurs niveaux d’annotation :
1. L’annotation syntaxique
Localisation des régions d’intérêts
2. L’annotation fonctionnelle
Attribution d’une fonction
3. L’annotation relationnelle
Mise en évidence de relation entre annotationANNOTER UN GÉNOME EUCARYOTE : THÉORIE
Les différents éléments que l’on peut annoter
Gènes Séquences Séquences de régulation
répétées
ARNs non-codant En tandem Promoteurs
ARN ribosomique rRNA Microsatellites Enhancers
petits ARNs nucléaires snRNA Minisatellites Locus control regions (LCR)
petits ARNs nucléolaires snoRNA Satellites Isolateurs (Insulators)
ARNs de transfert tRNA Silenceurs (Silencers)
micro ARNs miRNA Régions d’attachement à la
« Transfrags » matrice (matrix attachment
region, MAR)
Gènes codant des Dispersées
protéines
Gènes fonctionnels Retrotransposons
Pseudogènes Retroposons
TransposonsANNOTER UN GÉNOME EUCARYOTE : THÉORIE
Annotation syntaxique
Structure classique d'un gène
5’ UTR CDS 3’ UTR
intron intron
CAAT TATA AUG T AAUAA
exon exon exon
Deux façon d'annoter un gène :
par expertise humaine : manuelle
par un ou des programme(s) informatique(s) : automatiqueANNOTER UN GÉNOME EUCARYOTE : THÉORIE
Annotation Manuelle
Concerne un nombre restreint de gènes
Projet avec une large communauté d'annotateurs
http://vega.sanger.ac.uk/index.htmlANNOTER UN GÉNOME EUCARYOTE : THÉORIE
Annotation Automatique
Nécessite des ressources informatiques importantes
Plus rapide, moins fiable
http://www.ensembl.org/info/about/species.htmlANNOTER UN GÉNOME EUCARYOTE : THÉORIE La réconciliation des informations est la différence majeure entre l'annotation manuelle et l'annotation automatique.
ANNOTATION SYNTAXIQUE: DIFFICULTÉS
Structure classique d'un gène
5’ UTR CDS 3’ UTR
intron intron
CAAT TATA AUG T AAUAA
exon exon exon
Mais un gène classique, cela n'existe pas !ANNOTATION SYNTAXIQUE: DIFFICULTÉS
Paradoxe de la valeur C (= taille du génome)
La mouche du vinaigre
La taille du génome d’une espèce Eucaryotes ne peut pas Drosophila melanogaster
être déduit de sa « complexité »
180 Mb
Le criquet marcheur
Podisma pedestris
18,000 Mb
Gregory, T.R. (2010). Animal Genome Size Database. http://www.genomesize.comANNOTATION SYNTAXIQUE: DIFFICULTÉS
Taille du génome et nombre de gènes : chez les Procaryotes
E. coli
M. genitalium
Graur & Li. Fundamentals of Molecular Evolution (1999)ANNOTATION SYNTAXIQUE: DIFFICULTÉS
Taille du génome et nombre de gènes : chez les Eucaryotes
45000
40000 P. tetraurelia
O. sativa
35000
A. thaliana
30000
Nombre de genes
V. vinifera
25000
M. musculus
C. elegans H. sapiens
20000 T. nigroviridis
15000 C. intestinalis
D. melanogaster
T. brucei
10000 N. crassa
L. major
T. melanosporum
S. cerevisiae
5000 P. falciparum
0
0 500 1000 1500 2000 2500 3000 3500 4000
Taille du génome (Mb)ANNOTATION SYNTAXIQUE: DIFFICULTÉS
Taille du génome et nombre de gènes : chez les Eucaryotes
45000
40000
40000 37500
Nombre de gènes
35000
30000 27900
26900
28000
24000 24000
25000 20600
20000
14000
15000 12500
11200 11000
10000
9000
10000 6000 5200
5000 2000
0
na
um
s
r
m
is
ns
li
na
a
is
li a
ns
is
a
ei
ae
te
lu
cu
ss
iv
al
eu
al
id
uc
lia
ie
re
na
as
ga
si
cu
ar
at
ic
ra
tin
ni
ov
id
ap
au
br
ha
vi
ip
og
do
.s
le
op
us
cu
.c
es
co
re
gr
.s
lc
.e
tr
.t
T.
O
an
eu
.m
N
tr
ce
ni
nt
is
fa
te
E.
A
H
C
X.
ps
el
.d
.i
M
T.
P.
P.
S.
.m
C
D
T.
DANNOTATION SYNTAXIQUE: DIFFICULTÉS
Structure exon/intron espèce dépendante
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature (2001) 409: 745-964
Distance Nombre moyen
% répétitions Taille moyenne
Espèces Phylum intergénique d'exons par gène
génomiques des introns (nt)
moyenne (nt) (% monoexoniques)
Paramecium tetraurelia Ciliés 6% 27 335 3,3 (20%)
Vitis vinifera Dicotyledons 14% 970 11970 6 (8%)
Tuber melanosporum Ascomycetes 61% 107 6917 3,9 (18%)
3 génomes annotés au GenoscopeANNOTATION SYNTAXIQUE: DIFFICULTÉS
Quelques caractéristiques du génome humain
Chr. 20 Chr. 21 Chr. 22
Taille du chromosome 63 Mb 48 Mb 51 Mb
Gènes codants connus 551 235 445
Gènes codants nouveaux 8 7 24
Pseudogènes 201 141 275
ARN non codant 189 69 105
Densité en gènes codants 8,9 g./Mb 5 g./Mb 9,1 g./Mb
http://www.ensembl.org/Homo_sapiens/
Densité en gènes variable selon les espèces et même
selon les chromosomes.
Distribution non homogène des gènes sur les
chromosomesANNOTATION SYNTAXIQUE: DIFFICULTÉS
Gènes chevauchants
268 kbANNOTATION SYNTAXIQUE: DIFFICULTÉS Epissage alternatif dans plus de 80% des gènes humains (Matlin et al., Nature Reviews 6 (5): 386–398) Promoteurs alternatifs Signaux polyadenylations alternatifs
ANNOTATION SYNTAXIQUE: DIFFICULTÉS
Pseudogène
Duplication génique
duplication
Forme active Forme inactive
Retro-transposition d'un ARNm
transcription AAAA (ARNm)
Reverse - transcription
AAAA (cDNA)ANNOTATION SYNTAXIQUE: DIFFICULTÉS
# of repeats total bp
primates 563 664160
rodents 466 487006
other mammals 347 243730
other vertebrates 52 53994
Drosophila 65 167423
Arabidopsis 98 275516
grasses 27 67789
RepeatMasker (Arian Smit & Phil Green)
Moins de 50% des séquences répétées du génome humain sont spécifiques aux primates.
Mais presque la totalité des séquences répétées de souris sont spécifiques aux rongeurs,
car elles sont plus actives et possèdent un taux de mutation plus élevé.ANNOTATION SYNTAXIQUE: DIFFICULTÉS
H. sapiens M. musculus D. melanogaster A. thaliana V. vinifera C. elegans P. tetraurelia
Taille (Mb) 3275 3420 169 136 485 103 72
Nombre de
21727 22732 13781 31281 26346 20224 39642
gènes
Nombre
moyen 25,7 17,1 5,2 5 6,3 8 3,3
exons/gene
Densité
moyenne en
6,6 6,6 81,5 230 54 196 551
gènes
(gènes/Mb)
Chez les Eucaryotes :
la taille d'un génome ne reflète pas le nombre de gènes
les régions intergéniques sont de composition et de taille variable selon les espèces
la structure des gènes varie selon les espèces, et parfois au sein d'une même espèce
Du point de vue de l'annotation automatique, la complexité d'un génome dépend de sa variabilité.
Plus un génome aura des structures géniques homogènes, plus l'annotation automatique sera
performante.ASSEMBLAGE ET ANNOTATION DE GÉNOMES
EUCARYOTES
1. Introduction à l’assemblage de génomes
2. Annoter un génome Eucaryote : théorie et réalité
3. L'annotation automatique au Genoscope
4. Projets de génomique EucaryotesL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Le processus d’annotation
Recherche des structures exons-introns sur des séquences issues d'un assemblage
dans le but de définir un ensemble de modèles de gènes de référence.
Data Distribution
Genome browser
Submission
Data distribution
Masking Data Collection Integration Post Annotation Analysis
Known repeats, Proteins mapping Gene models Functional Annotation
low/simple/tandem Blat/Blast/GeneWise prediction InterProScan, KEGG KO
repeats using Gmove
RNA-seq (reads, contigs) Paralogous/Orthologous
RepeatMasker/TRF
Star/Gmorse/oases/trinity Metabolic Pathway
Repeats ab initio Ab initio genes predictions KEGG
detection SNAP
RepeatScoutL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Le masquage des répétitions connues
Les séquences répétées contenues dans les séquences génomiques (contigs, scaffolds,
chromosomes) sont masquées suivant plusieurs méthodes:
Tandem Repeat Finder (G. Benson, Tandem repeats finder: a program to analyze DNA
sequences, Nucleic Acids Research, 1999, vol. 27, No. 2, pp. 573-580.) permet de
masquer les répétitions en tandem sur des séquences génomiques sans a priori sur
les motifs à détecter.
RepeatMasker (A.F.A. Smit, R. Hubley & P. Green RepeatMasker at
http://repeatmasker.org/ ) permet de masquer des répétitions connues (simple, basse
complexité, organisme spécifique) sur des séquences génomiques. Il utilise Repbase
(http://www.girinst.org/repbase/index.html) pour rechercher les répétitions connues. Il
peut également utiliser une banque de séquence de répétition définit par l’utlisateur.
RepeatScout (Price et al. http://bix.ucsd.edu/repeatscout/) permet d’établir un
catalogue exhaustif des séquences répétées (connues et inconnues) présentes au
sein de l'organisme étudié. Attention, les familles de gènes peuvent être masquées.
L’objectif est de masquer le maximum de régions répétées non codantesL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Data Distribution
Genome browser
Submission
Data distribution
Masking Data Collection Integration Post Annotation Analysis
Known repeats, Proteins mapping Gene models Functional Annotation
low/simple/tandem Blat/Blast/GeneWise prediction InterProScan, KEGG KO
repeats using Gmove
RNA-seq (reads, contigs) Paralogous/Orthologous
RepeatMasker/TRF
Star/Gmorse/oases/trinity Metabolic Pathway
Repeats ab initio Ab initio genes predictions KEGG
detection SNAP
RepeatScoutL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Prédictions ab initio de modèles de gènes
Annotations
Recherche de modèles de gènes à partir d'un ensemble de Prédictions
Alignements protéiques
paramètres statistiques définissant un gène.
Calibrage de ces paramètres à partir de :
Paramétrages
soit de séquences connus de gènes de l'organisme
soit d'alignements de cDNAs de l'organisme
L'échantillon utilisé pour calibrer les outils doit être Prédictions
représentatif du protéome.
SNAP (Ian Korf, BMC Bioinformatics 2004, 5:59)
Modèles de gènes
Augustus (Stanke, et al., BMC Bioinformatics 2006, 7, 62)L'ANNOTATION AUTOMATIQUE AU GENOSCOPE
L’alignement de séquences exprimées
Objectif
Mise en évidence de régions génomiques codantes par comparaisons de séquences
exprimées (transcrits, protéines) conservées.
Mise en œuvre
Alignement de séquences de cDNAs et/ou de protéines contre des séquences
génomiques, du génome d’intérêt, issues de l’assemblage. Les séquences exprimées et
les séquences génomiques n’appartiennent pas nécessairement à la même espèce.
Contraintes Génomique
Tailles des séquences
Volume des données
Temps de calculs
Sensibilité / Spécificité des alignements
Redondance des matches
Recherche des introns
Protéines
cDNAsL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Annotation par comparaison de séquences
1) recherche d’un gène (mRNA) de la même espèce et
100% homologue, complet.
Nucléotides
2) recherche d’un gène (mRNA) de la même espèce et
100% homologue, partiel.
3) recherche d’homologies avec des gènes (mRNA, protéines)
de la même espèce, ou d’espèces différentes Acides aminés
4) recherche de régions de conservation avec d’autres
génomesL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Outils d’alignement Rapidité Spécificité Sensibilité
BLAT
BLAST
-BLASTN ntdb ntquery
-BLASTP aadb aaquery
-BLASTX aadb ntquery
-TBLASTN ntdb aaquery
-TBLASTX ntdb ntquery
BLAST.2
Idem BLAST mais introduit des “ GAPS ”
FASTA
Smith-WatermanL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Exploitation du RNAseq pour l’annotation
Mapping
Assemblage
Blat
Oases | Trinity
est2genome
Prédiction de
Reads
modèles de
Illumina
gènes
Mapping Modèles
STAR GmorseL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Exploitation du RNAseq pour l’annotation : Genome Contigs
Mapping
Découpage
Le génome et les transcrits appartiennent au même
organisme alignements stringent
Le génome et les transcrits n’appartiennent pas au Localisation des séquences de
même organisme (différentes souches, même cDNAs sur le génomique par un
alignement rapide (BLAT)
clade,…) alignement moins stringent
Filtre (longueur, score)
Définition des bornes exons/introns
sur le génomique par un alignement
(est2genome)
Modèles de gènes
(partiel ou complet)L'ANNOTATION AUTOMATIQUE AU GENOSCOPE Construction de modèles de transcrits à partir de données RNA-Seq Objectif : annoter des génomes eucaryotes à partir de données de transcriptome issues de séquençage haut-débit (Solexa/Illumina ou Solid) Difficultés : Prédire une structure de gène avec des tags d’une 100aine de bases Aligner les tags qui tombent sur une jonction exon/exon
L'ANNOTATION AUTOMATIQUE AU GENOSCOPE Gmorse : Gene modelling using RNAseq
L'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Les protéines conservées Genome Protéines
Alignements de séquences protéiques
(UniProtKB) sur l'assemblage dans le but Découpage
d'identifier des ORFs conservées entre
espèces.
La quantité d'alignements dépend du niveau Localisation des séquences de
protéines sur le génomique par un
d'homologie entre les organismes présents alignement rapide (BLAT)
dans les banques et celui à annoter.
Filtre (longueur, score)
Définition des bornes exons/introns
sur le génomique par un alignement
(genewise)
Modèles de gènes
(partiel ou complet)L'ANNOTATION AUTOMATIQUE AU GENOSCOPE
UniProtKB (release 2015_04)
UniProtKB (http://www.uniprot.org/ ) est composé de 2 sections:
SwissProt qui contient des protéines annotées manuellement et expertisées.
TrEMBL qui contient des protéines annotées automatiquement et non expertisées.
SwissProt
Kingdom sequences (%)
Archaea 19340 (4%)
Bacteria 332062 (61%)
Eukaryota 180260 (33%)
Viruses 16546 (3%)
Eucaryotes
Kingdom sequences (%)
TrEMBL
Archaea 913804 (2%)
Bacteria 29096694 (62%)
Eukaryota 13910429 (30%)
Viruses 2236332 (5%)
Other 557256 (L'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Data Distribution
Genome browser
Submission
Data distribution
Masking Data Collection Integration Post Annotation Analysis
Known repeats, Proteins mapping Gene models Functional Annotation
low/simple/tandem Blat/Blast/GeneWise prediction InterProScan, KEGG KO
repeats using Gmove
RNA-seq (reads, contigs) Paralogous/Orthologous
RepeatMasker/TRF
Star/Gmorse/oases/trinity Metabolic Pathway
Repeats ab initio Ab initio genes predictions KEGG
detection SNAP
RepeatScoutL'ANNOTATION AUTOMATIQUE AU GENOSCOPE Intégration des ressources
L'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Gmove: Gene Modelling using Various Evidence
RNAseq alignments
Proteins alignments
Ab initio predictions
Creation of an oriented graph
Final transcript
Putative exons and introns
Select
candidate
transcript
Extract paths from the graph
Transcript candidates Predicted transcripts
ORFs findingL'ANNOTATION AUTOMATIQUE AU GENOSCOPE Intégration des ressources
L'ANNOTATION AUTOMATIQUE AU GENOSCOPE Mesure de la qualité des annotations
L'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Qualité d’une annotation automatique
Application de filtres sur les modèles de gènes :
règles de filtrage basées sur la structure et le
score des gènes
détection de domaines protéiques et élimination
des protéines annotées présentant des domaines
de transposons
Evaluation de la qualité des annotations par
l'observation de plusieurs indices : recouvrement avec
Plot de densité montrant la corrélation entre les
les cDNAs de l'organisme, structure des modèles GC3 des codons des gènes orthologues du riz et
de la banane.
(tailles des protéines, exons, introns, nombre moyen
d'exons/gène,…), expertise manuelle.
Utilisation d’annotations fiables d’espèces voisines.
On comparera ensuite les gènes orthologues et les
caractéristiques suivantes :
- la teneur en GC aux positions 2 et 3 des codons
- le nombre d’exons par gène
- la taille moyenne des CDS (coding sequence)
Example of comparison of exon numbersL'ANNOTATION AUTOMATIQUE AU GENOSCOPE
Les ressources informatiques disponibles pour le calcul
Linux AMD et Intel x86 64 bits CCRT : BULL >3000 cores and 3 nodes with
64 cores and 3To
normal: >1000 cores ; 8Go-16Go/core + Curie supercomputer (360
nœuds/32coeurs/128Go RAM))
xlarge : 2x 40 cores; 2To RAM Lustre 5Po (with 2Po on HDD)
1x 48 cores ; 2To RAM
1x 48 cores ; 3To RAM
NAS Netapp ~ 1PoSEQUENÇAGE ET BIOINFORMATIQUE
http://www.businessinsider.com/super-cheap-genome-sequencing-by-2020-2014-10?IR=TASSEMBLAGE ET ANNOTATION DE GÉNOMES
EUCARYOTES
1. Introduction à l’assemblage de génomes
2. Annoter un génome Eucaryote : théorie et réalité
3. L'annotation automatique au Genoscope
4. Projets de génomique EucaryotesPROJETS DE GÉNOMIQUE EUCARYOTES
Tuber melanosporum Pisum sativum
(truffle) Vitis vinifera (pea) Flickr/chaojikazu
(grape)
Brasssica napus
(seed rape)
Theobroma cacao
(Cacao)
Chondrus crispus
(red alga)
Oncorhynchus mykiss Quercus robur
Triticum sp
(trout) Rosa chinensis (oak)
(wheat)
(rosebush) Coffea
(coffee)
Musa acuminata
(banana)
Et beaucoup d’autres organismes….LE GÉNOME DU BANANIER
2012 Aug 9; 488: 213–217LE GÉNOME DU BANANIER
‒ Source d'alimentation pour de plus de 400 millions de
personnes des pays du Sud
‒ Exportée massivement vers les pays développés
‒ La variété exportée est sensible à de nombreux parasites (il
s'agit d'un clone stérile : impossibilité de faire des croisements
pour sélectionner des plantes résistantes)
banane sauvage (fertile): non
comestible car présence de grainesLE GÉNOME DU BANANIER Génome 523 Mb, 11 chromosomes, 36 542 gènes ● L'annotation des gènes pourra permettre l'identification de facteurs de résistance aux pathogènes ou de qualité des fruits. ● Le bananier est la première plante de sa classe botanique (les monocotylédones), à côté des céréales, pour laquelle un séquençage abouti a été obtenu. Il constitue à ce titre une référence pour étudier l’évolution des génomes.
LE GÉNOME DU BANANIER
Le bananier a connu trois épisodes de duplication complète du génome indépendantes de celles
constatées dans la lignée des graminées. WGD
céréales
La plupart des gènes issus de ces évènements de duplication
sont ensuite perdus, mais certains persistent et permettent
l’émergence de nouvelles fonctions biologiques, comme
certains facteurs de régulation (facteurs de transcription) qui
sont particulièrement abondants chez le bananier et
concourent à des processus importants dont la maturation des
fruits.
WGDLE GÉNOME DU COLZA
LE GÉNOME DU COLZA
Le colza, une espèce récente à fort potentiel de
diversification et d’adaptation
• Première oléagineuse cultivée en Europe en termes de surface.
Explosion de sa culture à partir du 20e siècle
• Famille des Crucifères, comme la moutarde, le chou, le chou-fleur, le
brocoli, le chou chinois, le navet…
• Apparition il y a quelques milliers d’années (ère post-néolithique) : fruit
du croisement interspécifique favorisé par l’homme de façon
involontaire entre le chou et la navette.
• L’espèce s’est rapidement diversifiée en plusieurs types : colza à huile,
navet suédois, chou frisé, rutabaga…
Le séquençage de son génome constitue une ressource unique pour
l’amélioration variétale:
teneur et composition en huile
résistance à des pathogènes
tolérance au froid
rendement
efficacité d’utilisation des nitrates dans le sol
Triangle de ULE GÉNOME DU COLZA Le colza: champion des duplications récurrentes des génomes par polyploïdie Le colza a accumulé au cours de son évolution 72 génomes ancestraux, résultat de nombreux cycles de polyploïdisation, faisant de son génome un des plus hautement dupliqués chez les plantes à fleurs (angiospermes). Ce phénomène récurrent, suivi par des restructurations du génome, a conduit à l’accumulation d’un grand nombre de gènes: 101 040.
PROJET TARA OCEANS
Etude de la biodiversité des
micro-organismes marins
Les océans couvrent les 3/4 de la surface
de la Terre
Le plancton représente 80% des
organismes unicellulaires sur Terre
Dans 1L d’eau de mer :
Nombre d’espèces Phylum
100 – 1000 Animaux
10 000 – 100 000 Protistes
1 – 10 millions Bactéries
10 – 100 millions Virus
Bloom planctoniquePROJET TARA OCEANS
CO2
Eléments minéraux O2 Evaporation
Phytoplancton
Zooplancton
Matière Minérale
LE PLANCTON Matière Organique
Petits Poissons
50% de l’O2 produit
70% du CO2 recyclé
Grands Prédateurs Décomposition
BactériesPROJET TARA OCEANS
Catalogue
d’espèces du
plancton marin et
leurs interactions
Corrélation climat /
plancton
Christian Sardet (CNRS, Villefranche-sur-Mer)PROJET TARA OCEANS
IMAGERIE OCEANOGRAPHIE
INFORMATIQUE GENOMIQUE
Collaboration internationale impliquant une 20aine de laboratoiresPROJET TARA OCEANS http://oceans.taraexpeditions.org/
PROJET TARA OCÉANS Acquisition des données Satellite et modèles : informations océanographiques À bord : échantillonnage et stockage, imagerie, mesures en temps réel. À terre : traitements, analyses et modélisation (biodiversité taxonomique, fonctionnelle)
PROJET TARA OCEANS 3 méthodes, plus de 27 000 prélèvements : - Les filets : 7 modèles de 5 à 690µm, de la surface jusqu’à 1000 m de profondeur - La pompe péristaltique : de 10 à 120m de profondeur, pompe l’eau qui est filtrée dans des tamis de plus en plus petits. - La rosette CTD : caractérise les masses d’eau : pression, température, azote, O2 fluorescence…
PROJET TARA OCEANS
GPSS(Gravity Plankton
Sieving System)
Pompe péristaltique
La rosette
FiletsPROJET TARA OCEANS http://oceans.taraexpeditions.org/
PROJET TARA OCEANS
http://oceans.taraexpeditions.org/PROJET TARA OCÉANS
Cultures Biodiversité
Non cultivable
Cellules Communautés
Isolées de cellules
Séquençage Traitements &
à haut débit stockage
informatique
-> Liste des
espèces
-> Liste des
fonctionsStratégie
Tara
Single
Samples Cell
Isolation
DNA, RNA
Cultures
extraction
Whole
Genome
Séquençage
Tags Amplification
rRNA
mRNA
DNA
Assemblage
Prédiction de gènes
Annotation fonctionnellePROJET TARA OCÉANS
Analyse d’un assemblage de metagénomes
DNA Contigs from TARA METAGENOME set 1
Virus Archeae Bacteria Eukaryota Ambiguous Not Assigned
100%
90%
80%
70%
60%
50%
40%
30%
20%
10%
0%
0.2um 5-20um 20-180um 180-2000um >2000um
17/10/2016R&D BIOINFORMATIQUE ET SÉQUENÇAGE
R&D Bioinformatic et Sequencing group
Laboratoires de Séquençage du
Genoscope (LS, LBioMeG)
Laboratoire d’Analyse des
Génomes Eucaryotes (LAGE)
Production / Evaluation Assemblage
Caroline Belser Carole Azema Dossat
Stefan Engelen Arnaud Couloux
Frédérick Gavory Simone Duprat
Aurélie Périn Léo D’Agata
Sabrina Davidas Sébastien Faye
Eidji Bord Benjamin Istace
Artem Kourlaiev François-Xavier Babin
Annotation
Corinne Da Silva
Benjamin Noel
Marc Wessner
http://www.genoscope.cns.fr/rdbioseq
Marion Dubarry
Fabien DutreuxVous pouvez aussi lire

















































