CONSTANCE ET VARIATION DU DNA REPLICATION ET MAINTIEN - UNIVERSITE CADI AYYAD, FACULTE DES SCIENCES SEMLALIA DEPARTEMENT DE BIOLOGIE BIOLOGIE ...
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
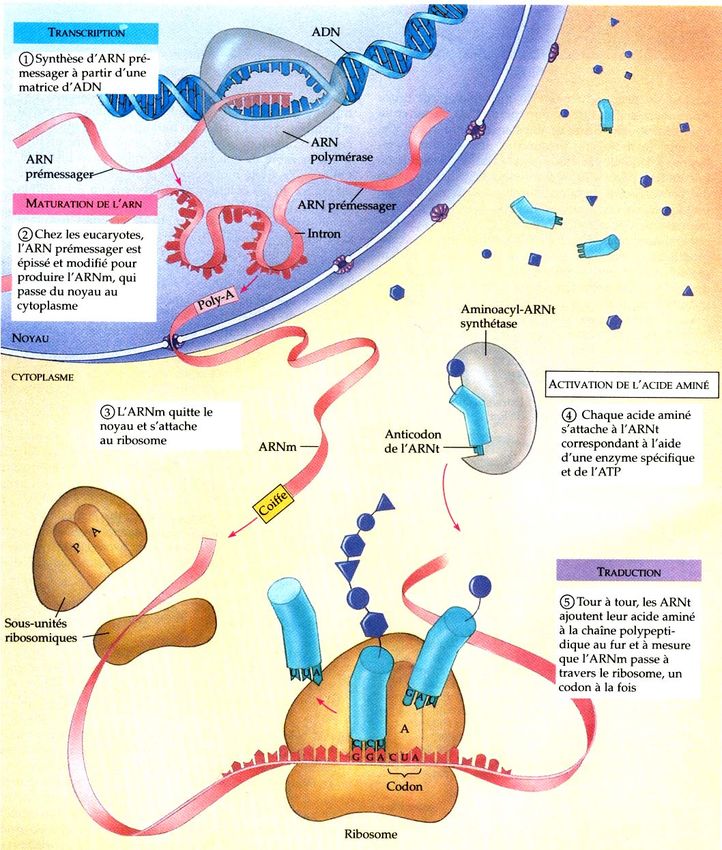
UNIVERSITE CADI AYYAD, FACULTE DES SCIENCES SEMLALIA
DEPARTEMENT DE BIOLOGIE
BIOLOGIE MOLECULAIRE
COURS S4 (PREMIERE PARTIE)
CONSTANCE ET VARIATION DU DNA
[ REPLICATION ET MAINTIEN ]
PROFESSEUR A. A. BENSLIMANE
2006
-0-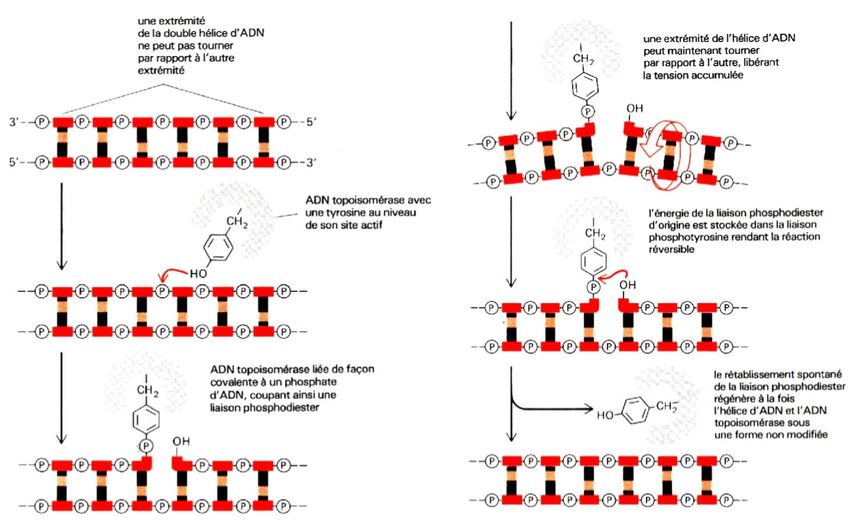
CONSTANCE ET VARIATION DU DNA
[ REPLICATION ET MAINTIEN ]
SOMMAIRE
A. CONSTANCE DU DNA 2
I. LA REPLICATION 2
1. LA REPLICATION CHEZ LES PROCARYOTES 2
α- EXPERIENCE DE MESELSON ET STAHL 3
β- SYNTHESE DE DNA IN VITRO 6
γ-REPLICATION IN VITRO 8
δ- OBSERVATIONS DE CAIRNS 10
ε- MECANISMES GENERAUX DE LA REPLICATION 11
INITIATION 12
DEROULEMENT DU DNA PARENTAL 14
ELONGATION 15
FINITION DU BRIN RETARDE 20
TERMINAISON 20
2. LA REPLICATION CHEZ LES EUCARYOTES 21
α- CYCLE CELLULAIRE 21
β- MULTIPLES POINTS D’INITIATION 22
γ- ORIGINES ET INITIATIONS 23
δ- VERS UN MODELE DE LA REPLICATION CHEZ LES 24
EUCARYOTES
ε- REPLICATION DES TELOMERES 27
ζ- MODIFICATION POST-REPLICATION DU DNA. 29
II. MAINTIEN DE L’INTEGRITE DU DNA ET REPARATION 30
1. LE MAINTIEN DE L’INTEGRITE DU DNA EST ASSURE PAR DES 33
SYSTEMES DE SAUVEGARDE
α- FIDELITE DE LA REPLICATION 33
β- SYSTEMES PREVENTIFS 34
2. LA REPARATION DU DNA EN DEHORS DE LA REPLICATION MET 34
EN JEU DES SYSTEMES MULTIPLES
α- REVERSION DIRECTE DU DOMMAGE 34
β- REVERSION EN DEUX PHASES 35
PHASE 1 : Détection, suppression de l’altération 35
PHASE 2 : Remplacement du DNA altéré 36
3. ANOMALIES DES SYSTEMES DE REPARATION CHEZ L’HOMME 37
-1-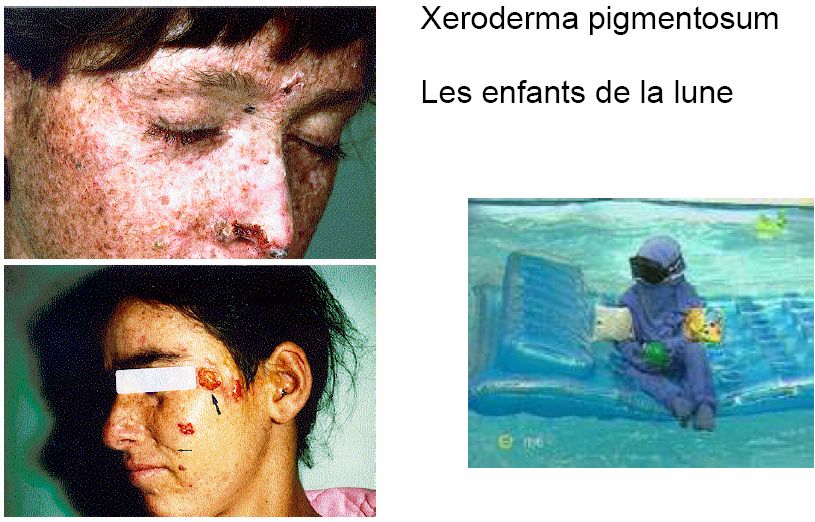
CONSTANCE ET VARIATION DU DNA
A.CONSTANCE DU DNA
La constance du DNA résulte de deux processus : la réplication et la réparation.
Les structures, propriétés et localisations des acides désoxyribonucléiques ont été décrites en
SV3. Dans ce chapitre, nous étudierons d’abord la biosynthèse du DNA, appelée aussi
« réplication ». Puisque ce processus est beaucoup mieux connu chez les procaryotes que chez
les eucaryotes, nous commencerons par l’étudier chez les premiers, puis le développerons
chez les seconds. Nous envisagerons ensuite les phénomènes de réparation des molécules de
DNA. Celles-ci doivent en effet durer aussi longtemps que les cellules qui les contiennent.
Elles doivent par conséquent être réparées lorsqu’elles sont abîmées.
I. LA REPLICATION
La réplication perpétue l’information génétique : Au cours de la vie de la cellule, d’une
division mitotique à la suivante, le DNA doit être dédoublé pour que chaque cellule fille
reçoive un génome complet identique à celui de la cellule mère de départ.
Les mécanismes qui conduisent à la réplication du DNA ont d’abord été élucidés chez les
procaryotes. La caractérisation de nombreux mutants conditionnels a permis de démontrer
presque parfaitement la totalité du mécanisme.
Le système est moins bien connu chez les eucaryotes. Cependant les résultats accumulés
montrent que les mécanismes y sont très semblables.
1. LA REPLICATION CHEZ LES PROCARYOTES
En même temps que leur modèle, Watson et Crick [1953] proposaient des implications
fondamentales à la structure secondaire du DNA. Cette structure secondaire montre
clairement qu'il existe deux "copies" de l’information génétique codée : l'une en positif, l'autre
en négatif découlant l'une de l'autre par complémentarité des bases.
On verra plus tard par quels mécanismes le DNA stocke toute l'information nécessaire au
développement de l'organisme et par quels mécanismes cette information est exploitée pour la
synthèse de l’ensemble des protéines cellulaires.
Un modèle de synthèse "semi conservatif" du DNA reposant sur cette observation a été
proposé et s'est avéré exact.
Lorsque l'information est transmise, d'une cellule
mère à deux cellules filles, les copies (positif et
négatif) doivent être représentées dans les deux
nouvelles cellules :
le modèle propose que chaque copie conserve un
des deux éléments du modèle (d'où l'expression
semi conservative associée à cette duplication), le
négatif ancien et un positif nouvellement synthétisé
va être hérité par une cellule fille, le positif ancien
et un négatif nouvellement synthétisé étant hérité
par l'autre cellule fille.
-2-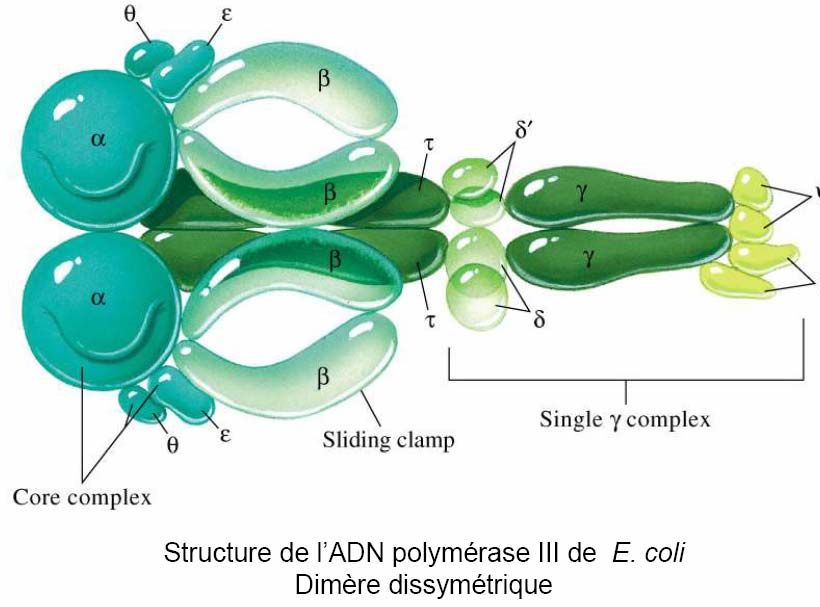
En moins d'un an, Meselson et Stahl concevaient une expérience restée célèbre pour vérifier
ces prédictions.
α- EXPERIENCE DE MESELSON ET STAHL
Introduction :
Cette expérience date de [1958]. Elle permet de démontrer le caractère semi-conservatif de la
duplication de la molécule de DNA chez les bactéries. Cette expérience a pu être réalisée
grâce à plusieurs mises aux points techniques :
1 - Meselson et Stahl mettent au point une technique d'obtention de gradient
de densité par centrifugation (centrifugation isopycnique). En utilisant du
chlorure de Césium de densité moyenne 1,72, ils obtiennent après 24h de
centrifugation à grande vitesse (ultracentrifugation) un gradient de densité
(de 1,70 à 1,75), gamme qui englobe la densité moyenne du DNA (1,710).
Les densités "ρ" sont exprimées en g/cm3.
2 - Ils cultivent les bactéries dans un milieu dans lequel les substances
organiques utilisées comme source d'azote contiennent de l'azote lourd
(15N). Au cours de la culture, toutes les molécules azotées et en particulier le
DNA contiennent une forte proportion d'azote 15N. Le DNA "lourd" a une
densité de 1,724 et peut être distingué du DNA "léger" (1,710). Le DNA
"léger" provient de bactéries cultivées dans un milieu contenant de l’azote
14
N.
3 - Ils mettent également au point une méthode qui permet de synchroniser
la division des bactéries pendant quelques générations.
Le problème à résoudre :
Depuis Watson et Crick [1953], on sait que le
DNA est une molécule formée de deux brins
antiparallèles, formant une double hélice. Dès
leur publication originale sur la structure du DNA,
Watson et Crick ont proposé que cette double
hélice puisse s'ouvrir, permettant ainsi la synthèse
de nouveaux brins, complémentaires des brins
originaux.
Le DNA peut ainsi servir de matrice à sa propre
réplication, étape essentielle du cycle cellulaire.
Cette duplication du DNA permet de passer de
chromosomes à une chromatide à des
chromosomes possédant deux chromatides
identiques portant la même information génétique.
Lors de la mitose, ces deux chromatides sont
séparées : chaque cellule-fille héritant d'une
chromatide de chaque chromosome. On obtient ainsi deux cellules possédant la même
-3-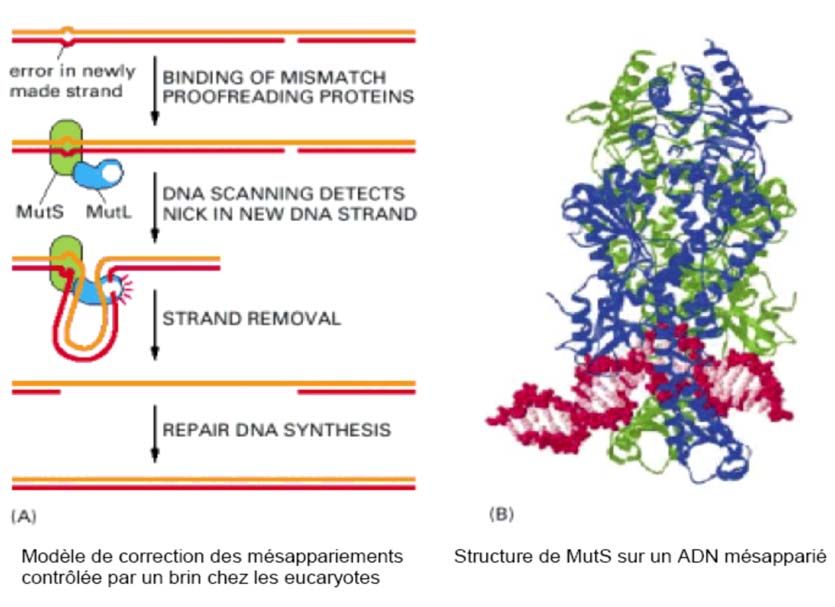
information génétique que la cellule-mère.
Le problème qui se posait à Meselson et Stahl était alors de comprendre comment se réalisait
cette réplication : selon quelles modalités passe-t-on d'une molécule de DNA formée de deux
brins à deux molécules de DNA bicaténaires identiques ?
Les hypothèses
Pour expliquer la duplication d'un DNA bicaténaire, trois modèles (a, b et c) ont été proposés.
Ces modèles se basent tous sur l'utilisation de la molécule de DNA "mère" comme matrice
pour sa réplication, mais selon des modalités différentes :
a- On dissocie les deux brins de la
molécule de DNA biacténaire
"mère". Chaque brin sert donc de
matrice à la synthèse d'un brin
complémentaire, l'ensemble
reformant une molécule de DNA
bicaténaire. Chaque nouvelle
molécule "fille" ne conserve donc
que la moitié de la molécule "mère".
b- A partir d'une molécule de DNA
bicaténaire "mère", on forme une
nouvelle molécule de DNA
bicaténaire. On garde donc ici une
molécule "mère", non modifiée (elle
est donc conservée), tout en "créant"
une nouvelle molécule ("fille").
c- On ne conserve aucun brin intact. La
copie se réalise par fragments
dispersés dans l'ensemble du DNA,
permettant de former les deux
molécules de DNA bicaténaires
"filles".
Matériel biologique
Le matériel expérimental est une cellule procaryote : Escherichia coli. Les bactéries sont en
effet les cellules présentant le plus haut rendement synthétique et dans des conditions standard
se divisent environ toutes les trente minutes ce qui va permettre d'accéder à plusieurs
générations et donc à plusieurs cycles de réplication de l'ADN en un temps raisonnable.
Expérience et résultats observés
Des bactéries cultivées depuis longtemps en présence de molécules azotées 15N sont repiquées
sur un milieu permettant la synchronisation des divisions et contenant des molécules azotées
14
N. Des fractions sont prélevées après différents temps correspondant à 1, 2, 3, ... divisions.
Le DNA est extrait, placé dans la solution de chlorure de Césium et centrifugé à 100.000 g
pendant 24h. La position des DNA est repérée par une mesure de la densité optique
-4-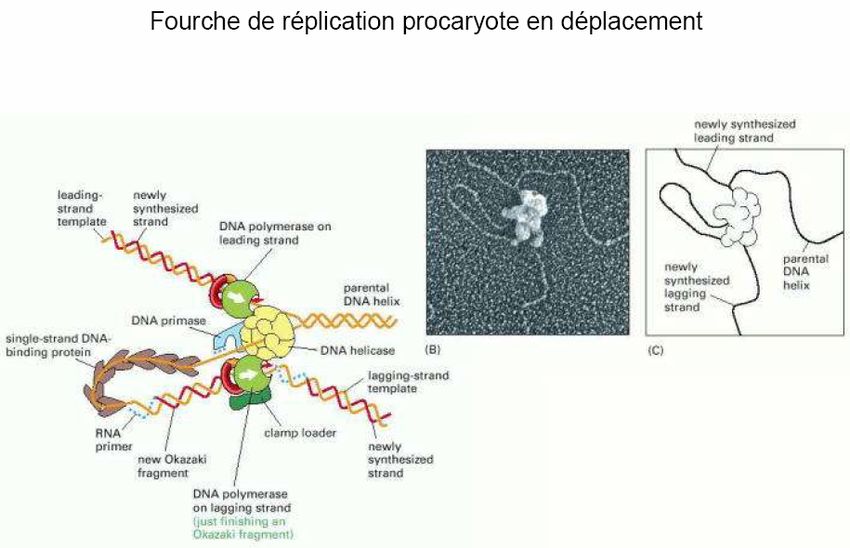
Position des
différentes bandes de
DNA14N
DNA au cours du
DNA hybride temps.
DNA15N Les chiffres donnent le
nombre de divisions
(ou générations)
Après 1 génération : tout le DNA est hybride (du point de vue de sa densité). Il n'y a plus de
DNA 15N. Ensuite, le DNA hybride disparaît progressivement au profit de DNA "léger" (14N).
L'expérience de Meselson et Stahl montre donc la présence d'un DNA hybride au bout d'une
génération cellulaire. Or, qu'attend-on pour les trois modèles proposés ?
(a) Modèle semi-conservatif (b) Modèle conservatif (c) Modèle dispersif
DNA hybride
(molécules formées DNA lourd (15N)
DNA hybride
d'un brin lourd et d'un et
(dispersif)
brin léger) DNA léger (14N)
On peut donc, dès cette première observation, rejeter le modèle conservatif (b).
Après deux générations cellulaires : Meselson et Stahl observent la présence de DNA
hybride et de DNA léger. Ceci permet de conclure quant aux deux modèles restants :
(a) Modèle semi-conservatif (c) Modèle dispersif
DNA hybride
Et DNA hybride
DNA léger
En conclusion :
Seul le modèle semi-conservatif permet d'aboutir expérimentalement aux résultats.
L'expérience de Meselson et Stahl permet donc de mettre en évidence le fait que la réplication
se réalise selon un mode semi-conservatif.
Cette conclusion a été depuis confirmée par des études plus précises, pour aboutir au modèle
actuel de fonctionnement de la réplication.
-5-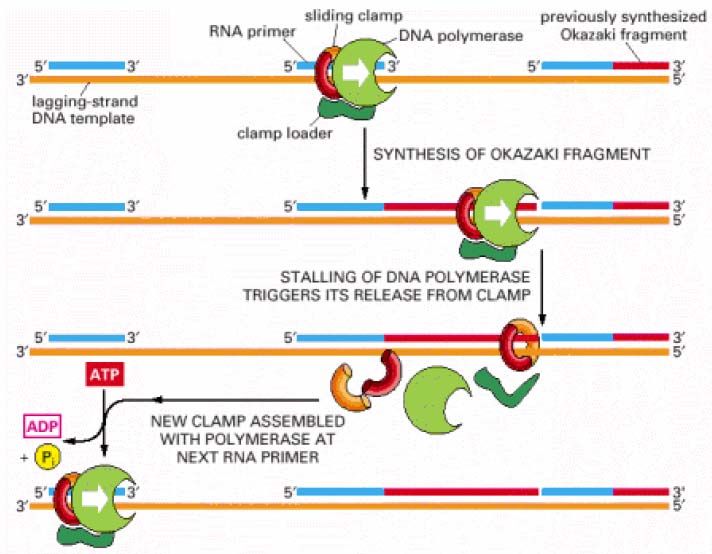
DNA parental
Brins néosynthétisés
0
1
2
Représentation schématique de la population de fragments de DNA au cours des générations.
Après 1 génération tout le DNA est "hybride" et constitué d'un brin "lourd" 15N et d'un brin "léger" 14N.
Quelques points importants de cette expérience sont à noter : Tout d'abord le fait qu'il est
nécessaire de séparer les DNA sur un gradient permettant de mettre en évidence leurs très
faibles différences de densités; une "simple" centrifugation ne suffit pas. L'utilisation d'un
gradient de Chlorure de Césium est donc un point fondamental du protocole. De même, ces
observations n'ont été possibles que parce que Meselson et Stahl avaient réussi à obtenir des
populations de bactéries synchrones (pendant quelques générations).
Très rapidement, plusieurs travaux remarquables confirment le mode de réplication du DNA
et laissent entrevoir la complexité du contrôle génétique de cette biosynthèse.
Dans les années [1958 à 1968], Kornberg réalise une première synthèse d'ADN in vitro et
Cairns "visualise" la réplication en microscopie électronique.
β- SYNTHESE DE DNA IN VITRO
Les séries d'expériences réalisées par Kornberg et son groupe [1958] préfigurent la génétique
moléculaire moderne et méritent que l'on s'y arrête.
-6-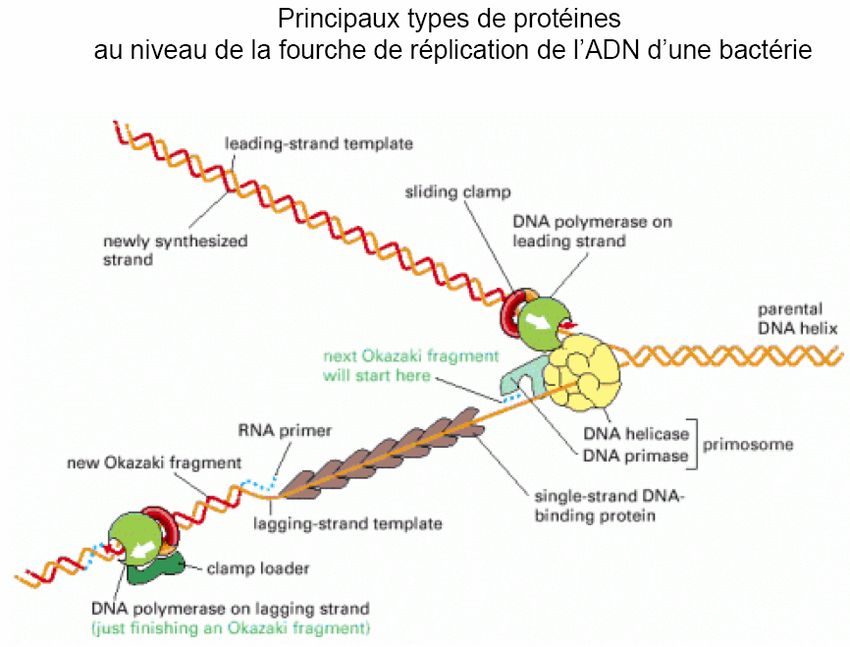
Le mode semi-conservatif de la synthèse du DNA implique les éléments suivants :
• - une molécule de DNA double brin capable de servir de modèle ou de matrice,
• - des désoxyribonucléotides précurseurs de la chaîne nouvelle,
• - une enzyme : DNA-polymérase, capable de relier ces précurseurs, cette enzyme
(hypothétique pour l'instant) est fondamentale car non seulement elle devra assurer la
liaison covalente (5'-3' phosphodiester) entre les nucléotides mais elle devra aussi être
capable de "choisir" ceux-ci en fonction du modèle présent selon la règle
d'appariement des bases.
Pour purifier et étudier cette enzyme, Kornberg a mis au point un système de synthèse in vitro
à partir d'extraits d'abord assez grossiers d'E. coli. Comment évaluer de tels systèmes ?
Comment prouver qu'une synthèse a bien lieu in vitro ? Comment distinguer le DNA
néosynthétisé de celui qui est obligatoirement présent dans l'extrait comme modèle ?
Kornberg va lui aussi faire appel à des marqueurs isotopiques : des nucléotides comportant
des phosphores 32 radioactifs (32P), si une synthèse a lieu, elle fera appel à ces précurseurs
radioactifs et le polymère résultant sera "marqué", sera radioactif et facilement repérable.
D'après l'analyse des nucléotides libres présents dans le cytoplasme, Kornberg décide de
choisir des nucléotides triphosphorylés en 5' alors que les constituants du DNA sont
monophosphorylés et que bien souvent, l'hydrolyse de polynucléotides produit des
mononucléotides phosphorylés en 3' ! On verra que sans cette décision, l'expérience était
vouée à l'échec : la cellule utilise effectivement des nucléotides 5' triphosphorylés et l'énergie
fournie par la libération du pyrophosphate.
Dans son mélange réactionnel, Kornberg dispose de DNA modèle, de précurseurs naturels, de
DNA polymérase active (il l'espère), auquel il ajoute des précurseurs radioactifs. Après la
réaction, la radioactivité se trouvera partagée entre le DNA éventuellement synthétisé in vitro
(en incorporant des monomères marqués) et l'excédent de précurseurs qui n'ont pas été
incorporés. Il est donc essentiel d'éliminer tous ces précurseurs libres pour attribuer de la
radioactivité à une macromolécule. En pratique, les macromolécules sont précipitées par
adjonction d'un acide organique et les petites molécules "acido-solubles" (y compris les
précurseurs radioactifs) sont éliminées par centrifugation. Le culot, après plusieurs lavages,
contient les macromolécules (y compris le DNA) débarrassées de tout précurseur non
incorporé dans la chaîne.
En suivant la stratégie exposée et ses contraintes, Kornberg fut capable de trouver quelque
radioactivité dans des fractions acido-précipitables. Radioactivité qui, à l'époque ne dépassait
guère le seuil de confiance des compteurs, mais Kornberg y croyait !
Plusieurs équipes, partant de kilogrammes de pâte d'E. coli, à l'aide de méthodes d'analyse
biochimique classiques de nos jours mais que l'on découvrait à l'époque, ont peu à peu
concentré l'activité de la DNA polymérase jusqu'à purifier cette enzyme qui fut nommée
"polymérase de Kornberg", plus connue de nos jours sous en tant que DNA polymérase I.
Le bilan (provisoire) de ces expériences est le suivant :
DNA DNA + DNA
-7-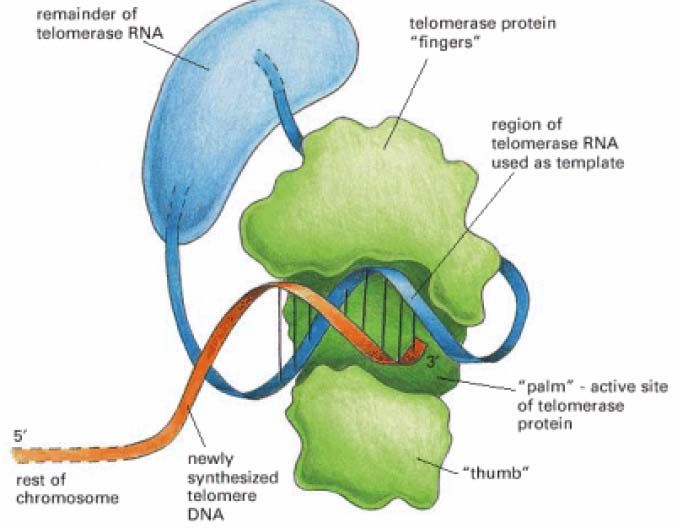
Par la suite, l’isolement de mutants conditionnels thermosensibles chez E. coli a permis
d’identifier les protéines jouant un rôle primordial dans la réplication. C’est ainsi que l’on a
pu montrer que l’enzyme « réplicative » n’était pas la DNA polymérase I, mais la DNA
polymérase III.
γ-REPLICATION IN VITRO
L'expérience décrite ci dessus prouve qu'une synthèse de polydésoxyribonucléotide est
réalisable in vitro mais ne prouve pas que le DNA synthétisé soit conforme au modèle de
départ ni que la synthèse soit une réplication semi-conservative.
La suite du travail va consister à tenter la synthèse in vitro d'un DNA "biologiquement actif",
l'activité biologique la plus facile à détecter étant, à l'époque, la capacité d'infection d'un DNA
de bactériophageФΧ 174. Les bactériophages utilisent essentiellement, pour leur réplication,
les protéines de la machinerie de leur hôte (E. coli dans notre cas)
Le modèle choisi (phage ФΧ 174) correspond à une molécule circulaire d'environ 5000
nucléotides seulement ; Nucléotides et non pas paires de nucléotides car il s'agit, pour la
particule phagique d'un DNA simple brin que nous appellerons le brin +. Le changement d'un
seul de ces nucléotides rend la molécule inactive (non infectieuse). La réalisation d'une copie
infectieuse in vitro va préfigurer la technologie du DNA recombinant : [1968 :Goulian,
Kornberg et Sinsheimer].
• - In vivo, la première étape de l'infection par ce bactériophage simple brin est la
synthèse d'un brin complémentaire pour réaliser une forme circulaire double brin à
partir de laquelle seront reproduits des brins + qui assureront la descendance phagique.
• - Un premier problème se posa pour la synthèse in vitro d'un brin - : la DNA
polymérase purifiée ne peut qu'attacher l'extrémité 5' d'un nucléotide à l'extrémité 3'
d'une chaîne en cours de synthèse, elle ne peut relier des polynucléotides et donc ne
peut pas réaliser la liaison phosphodiester qui permet de circulariser un brin de DNA.
Le problème a été résolu par la purification d'une enzyme qui, in vivo, remplit cette
fonction : c’est la DNA ligase dont nous aurons souvent l'occasion de parler.
• Le système de réplication in vitro va donc comprendre :
o des molécules de DNA purifiées de ФΧ 174 (brin +)
o les 4 désoxyribonucléotides
o DNA polymérase (extraits de E. coli)
o la ligase
• Problème 2 : en principe, ce système ne peut assurer que la synthèse de brins moins
circulaires complémentaires du brin plus. le brin - synthétisé in vitro n'est pas
infectieux, seul un brin + peut l'être. Il va donc falloir recommencer une synthèse in
vitro en utilisant les brins - comme modèles
• - Problème 3: comment séparer les brins - des brins + ? Ce problème a été surmonté
par l'utilisation d'un précurseur particulier à la synthèse du DNA : la 5-bromodésoxy
uridine, cet analogue de nucléotide est utilisé par la cellule comme de la thymidine (il
sera apparié aux résidus A de la matrice) mais le brome va "alourdir" la molécule de
DNA qui utilise ce précurseur. La différence de densité est suffisante pour permettre la
-8-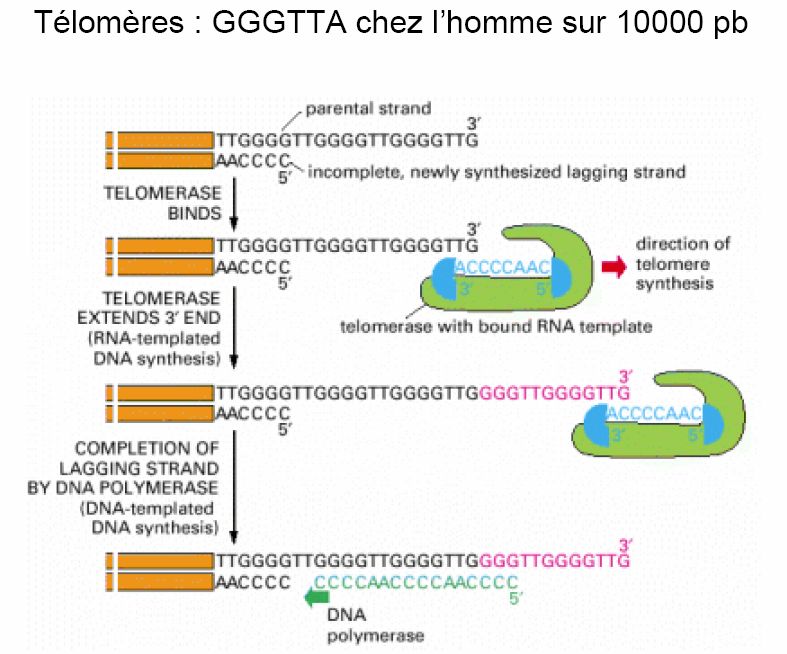
séparation de brins + (comportant de la thymidine) de brins - (comportant de la
bromodésoxyuridine) par ultracentrifugation sur un gradient de densité de CsCl.
Cette stratégie a permis de synthétiser des molécules qui vont s'avérer infectieuses : aucune
erreur sur 5000 nucléotides assemblés in vitro !
Remarque 1 : Il est important d’observer que la réplication dans cette expérience est menée à
bien en deux étapes successives et non pas simultannées, si bien qu’il n’est pas possible de la
considérer comme un modèle du mécanisme biologique de la réplication du DNA.
Remarque 2 : en fait, ce genre d’études a également permis de mettre en évidence
l’intervention d’autres éléments cellulaires dans le processus de la réplication tels que la DNA
polymérase III et la primase, responsable de la synthèse non seulement de l’amorce RNA à
l’origine de la réplication du phage, mais aussi des fragments d’Okazaki qui seront abordés et
décrits un peu plus loin.
-9-δ- OBSERVATIONS DE CAIRNS
Cairns [1962] a été le premier à observer un chromosome entier d'E. coli en cours de
réplication.
Il a associé des techniques de marquages isotopiques et d'autoradiographie suivis
d'observation en microscopie électronique. Après avoir cultivé des bactéries dans un milieu
contenant de la thymidine tritiée à faible activité spécifique, pendant un temps dépassant la
durée du cycle, il met au point une méthode de lyse de la cellule permettant de libérer le DNA
directement sur une grille de microscopie électronique, en minimisant les risques de cassures
mécaniques de la molécule.
La préparation est recouverte d'une émulsion photographique et après exposition et
développement, l'examen révèle des grains d'argent le long de la molécule de DNA. Ces
premières observations ont montré la circularité du chromosome d'E.coli, forme qui s'avérera
très répandue chez les procaryotes, les virus et le DNA des organites (mitochondries et
chloroplastes) des cellules eucaryotes.
Dans un second temps, Cairns a effectué des marquages plus courts et a déduit des images
obtenues que la réplication commence en un seul point du chromosome bactérien à partir
duquel se forme un « œil » qui ne cessera de grandir jusqu’à ce que deux chromosomes
bactériens soient obtenus.
Fourches de réplication
bidirectionnelle
Origine de réplication
Deux copies du chromosome
bactérien original
Fourches de
réplication
Point de
départ de la
réplication
- 10 -Un peu plus tard, d'autres chercheurs ont ajouté à un marquage long par la thymidine tritiée à
faible activité spécifique un marquage très bref par de la thymidine tritiée à forte activité
spécifique. Après autoradiographie, l'intensité des grains permet de distinguer les deux
marquages. On observe alors, des sortes de "bulles". Les figures matérialisées par les grains
d'argent seront appelées « fourches de réplication ».
FOURCHES
L'interprétation de ces figures va avoir un impact considérable :
• D'après l'observation de ces "fourches", il est clair que la réplication se fait
simultanément à partir des deux brins anciens.
• Puisque l'on observe deux de ces fourches, c'est que la réplication est bidirectionnelle.
• Si la réplication est bidirectionnelle c'est qu'il existe une "origine de réplication".
Cette notion n'est pas que topographique, on verra qu'effectivement, seule une
séquence précise de ces molécules circulaires permet le démarrage de la réplication.
Un élément quelconque d'un génome, naturel ou obtenu par génie génétique, ne pourra être
répliqué (et donc transmis à une descendance) que s'il possède une origine de réplication, il
sera alors considéré comme un "réplicon".
Le système enzymatique responsable de la réplication fonctionne donc à partir du point
d’initiation ou « origine de réplication » au niveau de deux fourches migrant en sens inverse
et se rejoignant en un point opposé du site d’initiation appelé « terminus ».
ε- MECANISMES GENERAUX DE LA REPLICATION
Tout modèle de système de réplication doit apporter une solution à deux types de problèmes :
1- Des problèmes d’ordre topologiques, car les structures de condensation du DNA
(boucles ou domaines surenroulés du chromosome bactérien) ont introduit des
supertours négatifs dans le DNA. D’autre part les deux brins du DNA sont enroulés en
double hélice. Il est donc impératif pour que la totalité du DNA soit répliquée, que les
brins soient progressivement séparés.
2- Des problèmes de synthèse semi-conservative posés par l’orientation antiparallèle des
deux brins.
Très rapidement, les études génétiques et biochimiques de la réplication ont montré que le
mécanisme est beaucoup plus complexe que ne l'évoque la prédiction de Watson et Crick et
que ne le laissent supposer les expériences de Meselson et Stahl (qui ne rendent compte de
l'état du DNA qu'avant et après la réplication), de Kornberg (qui isole la réplication de son
contexte cellulaire) ou de Cairns (qui fixe une image instantanée).
- 11 -Tous les transferts d'information que nous allons étudier comportent trois étapes dans la
synthèse de molécules informatives : le début, la suite et la fin que l'on préfère appeler les
étapes d'initiation, d'élongation (de la macromolécule en cours de synthèse) et de
terminaison. Des mutants pour chacune de ces étapes ont permis de les étudier en détail, c'est
l'initiation qui représente certainement l'étape clé de la réplication.
INITIATION
La réplication d’un chromosome bactérien est étroitement liée au cycle de croissance. Chez E.
coli, l’origine de réplication se situe à l’intérieur du locus génétique « oriC » et elle est fixée à
la membrane cytoplasmique. L’oriC contient quatre sites de fixation (9 paires de bases
chacun) pour la protéine initiatrice « Dna A ». La synthèse de cette protéine est liée à la
vitesse de croissance.
Une fois le niveau critique de croissance atteint, la protéine Dna A forme un complexe, de 30
à 40 molécules, autour duquel le DNA oriC est enroulé. Chacune des 30 à 40 molécules se
fixe à une molécule d’ATP. L’ensemble de ce processus requiert le surenroulement négatif du
DNA ce qui facilite la fusion "désappariement" de trois séquences répétées de 13 pb riches en
AT qui s’ouvrent pour permettre la fixation de la protéine « Dna B ».
La Dna B est une DNA
hélicase (appelée également
déroulase) qui casse les
liaisons hydrogènes qui
unissent les 2 brins du DNA.
Pour effectuer ce travail, de
l’énergie est nécessaire, elle
est fournie par l’hydrolyse
d’ATP. En fait, il existe
plusieurs types d’hélicases
agissant de concert (Rep
protéines, hélicases II et III).
Les brins du DNA ainsi
séparés sont stabilisés sous
forme simple brin grâce à la
fixation des « protéines
SSB » (ou ‘single strand DNA
binding’). Ces protéines sont
des tétramères d’une masse
moléculaire de 74 kDa. Leur
fixation à la molécule de DNA
est un phénomène coopératif :
la fixation d’un premier
tétramère sur le DNA favorise
- 12 -la fixation de la protéine suivante par augmentation de l’affinité apparente, et ainsi de suite
jusqu’à ce que tout le DNA passé sous forme simple brin soit recouvert de protéines SSB qui
forment une sorte de manchon. Cette association rigidifie les deux brins du DNA, les
empêche de se réassocier et les protège des cassures.
La synthèse de DNA par les DNA polymérases ne peut se
faire que par élongation d’une amorce (primer). Cette
amorce, comme l’a montré Okazaki [1968], est synthétisée
par un complexe protéique appelé « primosome ». Il est
constitué d’une RNA polymérase appelée « primase » codée
par le gène « dnaG » et de plusieurs autres protéines qui
assurent entre autres la reconnaissance des sites ou doit être
synthétisée l’amorce RNA.
- 13 -DEROULEMENT DU DNA PARENTAL
Pour que la réplication procède de l’origine, les DNA hélicases doivent se déplacer le
long des brins matrices pour ouvrir la double hélice (500 à 1000 nucléotides par
seconde ; le DNA parental doit tourner à raison de 50 à 100 tours par seconde).
Cependant, dans une molécule de DNA circulaire fermée, l’enlèvement de tours
hélicoïdaux au niveau de la fourche de réplication conduit à l’introduction de tours
supplémentaires dans le reste de la molécule sous forme de surenroulements positifs.
Bien que le surenroulement négatif naturel du DNA compense partiellement ce fait, il
reste cependant insuffisant pour permettre la progression de la fourche de réplication.
Le surenroulement positif, conséquent à l’avancement des DNA hélicases, doit être relaxé
continuellement par l’introduction permanente de surenroulements négatifs. Ceci est assuré
par des topoisomérases de typeI et II appelées aussi « DNA gyrases ».
Hélicase
SSB Molécule parentale
Topoisomérase
Primosome
SSB
- 14 -Les inhibiteurs de la DNA gyrase, tels que l’acide oxolinique et la novobiocine, sont des inhibiteurs
efficaces de la réplication bactérienne et possèdent par conséquent une activité antibiotique.
ELONGATION
L’élongation d’un nouveau brin de DNA au niveau de la fourche de réplication est catalysée
par la « DNA polymérase III ». Celle-ci synthétise un brin complémentaire à partir de
l’extrémité 3’OH libre de l’amorce RNA en utilisant le DNA comme matrice. La synthèse se
fait donc dans le sens 5’→3’. Les protéines SSB sont chassées au fur et à mesure de
l’utilisation du brin matrice.
Les deux brins complémentaires au DNA parental doivent être synthétisés dans le sens
5’→3’. Par conséquent, la synthèse ne peut être continue que sur l’un des deux brins :
« le brin direct », « brin principal », « brin précoce » ou « brin avancé » par opposition au
« brin indirect », « brin secondaire », « brin tardif » ou « brin retardé » dont la synthèse
se fait de manière discontinue.
- 15 -La DNA polymérase III est une enzyme très complexe composée de plusieurs sous unités
codées chacune par des gènes de structure.
Il semble que deux molécules de polymérase soient associées au niveau du point de
réplication, chacune répliquant son brin, ce qui implique un repliement d’un des brins sur lui-
même. L’enzyme sur le brin matrice 3’→5’ (brin direct) synthétise en continu au fur et à
mesure du déroulement des brins du DNA parental. L’enzyme située sur l’autre brin (brin
retardé) synthétise du DNA de manière discontinue sous forme de petits fragments (1000 à
2000 pb) appelés fragments d’Okazaki. Le DNA est ensuite ouvert sur une nouvelle longueur
par les hélicases, une nouvelle amorce est synthétisée sur le brin 5’→3’, les synthèses
reprennent et ainsi de suite jusqu’à ce que la totalité du réplicon soit dupliquée.
- 16 -Le fait qu’il y ait deux polymérases dans un seul complexe confirme que les deux brins sont
synthétisés à la même vitesse. Les deux moitiés du dimère contiennent une « sous-unité α »,
la vraie polymérase et une « sous-unité ε » qui est une exonucléase correctrice de 3’→5’. La
correction permet le maintien d’une haute fidélité de réplication. Les « sous-unités β »
servent à clamper la DNA polymérase. Les sous-unités restantes dans chaque moitié sont
différentes (structure dissymétrique).
- 17 -- 18 -
Lorsque la DNA polymérase III introduit un nouveau nucléotide, le taux d’erreur est
d’environ 10-4 à 10-5 (1x erreur sur 104 à 105 nouveaux nucléotides insérés). Or le taux
d’erreur au niveau du DNA
néosynthétisé n’est que de
10-9. Cette divergence vient
du fait que les DNA
polymérases sont capables de
détecter les erreurs qu’elles
ont commises et de les
corriger immédiatement.
Cette activité de contrôle-
correction d’épreuve
« proofreading » des
polymérases résulte d’une
activité 3’ exonucléasique
que possèdent en plus ces
polymérases.
On pense actuellement que le processus est le suivant : la polymérase en action entoure le
DNA comme un gros manchon. Si la base qui vient d’être ajouté n’est pas la bonne, la
structure dans l’espace du DNA est modifiée. Cette modification augmente le volume du
DNA et bloque la DNA polymérase. Cet arrêt permettrait à l’activité exonucléasique de
s’exercer, d’où excision de la base indûment incorporée, permettant ainsi la correction de
l’erreur et le redémarrage de la polymérase.
- 19 -FINITION DU BRIN RETARDE
Les amorces sont détruites par la « RNase
H » qui a pour propriété de détruire
spécifiquement le RNA des hybrides RNA-
DNA. La lacune engendrée par l’action de
cette enzyme est comblée par l’action de la
« DNA polymérase I ». Enfin, une « DNA
ligase » effectuera la soudure du brin
discontinu ; les DNA polymérases n’étant pas
capables de créer une liaison phosphodiester
qu’entre une extrémité 3’OH libre et un
nucléoside triphosphate libre. La DNA ligase
de E. coli utilise le cofacteur NAD+ comme
source inhabituelle d’énergie.
TERMINAISON
Les deux fourches de réplication se
rencontrent au terminus approximativement à
180° de l’oriC. Les deux cercles résultants
restent interliés. Ils sont dissociés par la
« topoisomérase IV », une DNA
topoisomérase de type II qui joue ici un rôle
de démêlage.
- 20 -Modèle d’action d’une topoisomérase type II
2. LA REPLICATION CHEZ LES EUCARYOTES
Les données sont encore insuffisantes pour pouvoir proposer un modèle définitif. Peut-être
n’est-il pas le même chez tous les eucaryotes ? Les données actuelles permettent cependant
de proposer le modèle suivant :
α- CYCLE CELLULAIRE
Le DNA est répliqué uniquement pendant le phase (S : synthèse). Celle-ci est précédée par
(G1 : gap 1) et suivie par (G2 : gap 2). La mitose (M) suit (G2). Les cellules peuvent
également interrompre le cycle cellulaire : elles entrent en (G0). Certaines cellules comme les
cellules nerveuses différenciées y resteront toute la vie de l’individu.
POINT T (transition)
G0
~ 3 heures Division
~ 1 heure
POINT R (restriction) Quelques heures
Point de non retour à quelques jours
(division ou suicide
par apoptose)
Synthèse du DNA
et des histones
7 à 8 heures
(40 minutes chez la levure)
- 21 -Il existe dans le cycle cellulaire, deux points cruciaux de contrôle (2 check points) : R et T.
R : Le passage de la phase (G1) à (S) est régulé par des « cyclines» et des « protéines
kinases cycline dépendantes » (Cdk-2 et Cdk-4).
T : Le passage de la phase (G2) à (M) est régulé par la « cycline B », la (Cdk-1) et
l’intervention de « kinase-kinase » tels que CAK et (wee-1) et de phsphatase (cdc-25). Les
divers enzymes seront activés ou inactivés par phosphorylation ou déphosphorylation.
β- MULTIPLES POINTS D’INITIATION
Le mécanisme global de réplication chez les eucaryotes est comparable à celui des
procaryotes. Elle se fait de manière :
Bidirectionnelle
Complémentaire
Antiparallèle
Dans le sens 5’ → 3’
Avec des amorces RNA, fragments d’Okazaki, brin retardé et brin avancé
Dans la cellule Eucaryote, les chromosomes comportent des molécules linéaires d'ADN très
longues et il existe plusieurs origines de réplication par chromosome, également caractérisées
par des séquences précises.
En raison de la complexité de la structure chromatinienne, les fourches de réplication
eucaryotes se déplacent à environ 50 pb/seconde (10 à 20 fois moins vite que chez E. coli). A
cette vitesse, en utilisant seulement deux fourches, la réplication d’une molécule de 105 kb
(taille moyenne d’un chromosome de mammifère) prendrait environ un mois.
En fait, au lieu de n’avoir qu’un seul point d’initiation (un seul réplicon), comme c’était le cas
chez les procaryotes, la réplication chez les eucaryotes débute simultanément en plusieurs
points d’un même chromosome (50 000 à 100 000 réplicons dans une cellule de mammifère ;
400 chez la levure Saccharomyces cerivisiae).
Oeil de réplication
- 22 -γ- ORIGINES ET INITIATIONS
Chez les eucaryotes, la réplication d’un génome ne se fait pas d’un seul bloc. Des groupes
d’environ 20 à 50 réplicons (répartis en tandem) se lancent simultanément à des moments
définis de la phase S. Ceux qui répliquent au début de la phase S abondent en euchromatine
alors que ceux activés en fin de phase S contiennent essentiellement de l’hétérochromatine.
Le DNA centromérique et le DNA télomérique se répliquent à la fin.
Réplication décalée
dans le temps et dans l’espace :
euchromatine d’abord
puis hétérochromatine.
(Brèves incorporations du BrdU à
différents stades de la phase S)
Les séquences correspondant aux origines de réplication chez les mammifères n’ont pas
encore été bien définies.
Les origines individuelles de réplication chez la levure (Saccharomyces cerivisiae) ont été
clonés en plasmides procaryotes. Puisqu’ils permettent à ces mêmes plasmides de se répliquer
dans la levure (un eucaryote), ils ont donc été appelé « ARS » : « séquence de réplication
autonome ». La longueur minimale du DNA qui supportera la réplication est uniquement de
11 pb bien que des copies supplémentaires de cette même séquence soient requises pour une
efficacité optimale. La séquence consensus des « ARS » est la suivante :
[A/T]TTTA[T/C][A/G]TTT[A/T]
Origines de réplication chez S.cerevisiae
- 23 -Cette séquence est liée par un complexe de reconnaissance de l’origine « ORC » ou « origin
recognition complex » qui, une fois activé par les Cdk, permet l’ouverture du DNA pour la
réplication.
S. cerevisiae
petit chrom os om e : 180 gènes ; 9 origines de ré plication
Contrairement aux procaryotes, les réplicons eucaryotes peuvent se répliquer seulement une
fois par cycle cellulaire. Une protéine appelée « facteur d’habilité », indispensable pour
l’initiation, est inactivée après son utilisation. Cette protéine ne peut avoir accès au DNA que
lorsque l’enveloppe nucléaire est dissociée à la mitose.
δ- VERS UN MODELE DE LA REPLICATION CHEZ LES EUCARYOTES
Une comparaison succincte avec le modèle procaryote est présentée sur le tableau suivant :
Le DNA est déroulé au niveau de l’origine de réplication grâce à au moins une topoisomérase,
une hélicase et au facteur « RF-A ».
Les nucléosomes sont déroulés en amont des fourches de réplication puis reconstitués en aval.
De nouveaux nucléosomes sont également assemblés exclusivement à partir de nouvelles
- 24 -molécules d’histone. Les
nucléosomes, nouveaux et
préexistants sont vraisemblablement
répartis équitablement entre les deux
DNA-fils. Durant la phase S, le
doublement du DNA est
parallèlement accompagné du
doublement du contenu en histones
de la cellule.
Addition de nouvelles histones en
arrière de la fourche de réplication
CAF : facteurs d’assemblage de la
chromatine
Sur le brin retardé, la « polymérase
α-primase » interagit avec le facteur
RF-A et synthétise une amorce RNA
d’une longueur d’une dizaine de
nucléotides. Cette amorce est ensuite
allongée grâce à l’activité DNA-
polymérasique de polymérase α
associée au « facteur RF-C » (une
vingtaine de désoxynucléotides). Le
fragment d’Okazaki est ainsi amorcé.
L’association du « PCNA »
(Proliferating Cell Nuclear Antigen)
et d’ATP provoque l’arrêt de la
polymérase α et permet la fixation
de la « polymérase δ » qui reconnaît
l’extrémité 3’OH néosynthétisée et
démarre alors la réplication.
La polymérase α libérée est transloquée avec ses protéines accessoires sur le brin direct où la
réplication est amorcée suivant le même principe.
Le brin direct est répliqué en continu alors que la synthèse s’arrête sur le brin retardé environ
250 nucléotides plus loin. Un nouveau fragment d’Okazaki est ensuite initié et ainsi de suite.
La « polymérase β » et « ligases » se chargent de la finition du brin retardé après excision des
RNA amorces.
On ne sait pas le rôle joué par la « polymérase ε ». Il a été proposé que le brin direct soit
synthétisé par cette polymérase mais ceci reste l’objet de controverses.
- 25 -RF-A
ε- REPLICATION DES TELOMERES
Dans un DNA circulaire, le remplacement des amorces RNA par du DNA ne pose pas de
- 26 -problème : le mécanisme de type réparation suffit (RNaseH, DNA polymérase I, ligase chez
les procaryotes).
Il n’en est pas de même avec les molécules de DNA linéaire où le retrait du RNA aux
extrémités des brins directs laisserait un trou qui ne peut être comblé : les DNA polymérases
ne fonctionnent que dans le sens 5’→3’. Ceci implique que si aucun mécanisme spécifique
n’avait été mis en place, la taille du génome ne cesserait de diminuer à chaque réplication.
Chez certains virus, comme l’adénovirus, la solution est apportée par une protéine qui se
fixe à l’extrémité du DNA lors de l’initiation de la réplication. Dans ces cas très particulier, il
n’est pas synthétisé de RNA amorce aux extrémités, c’est l’hydroxyle d’une sérine ou d’une
thréonine qui fait fonction de 3’OH et qui permet l’initiation de la réplication.
Chez les eucaryotes, pour surmonter cet obstacle, les extrémités des chromosomes ou
« télomères » sont constituées de centaines de séquences riches en G, non codantes, répétées
en tandem (250 à 1500 copies) avec l’extrémité en 3’ surplombant l’extrémité 5’.
Une enzyme appelée « télomérase » contient une petite molécule de RNA (c’est une
ribonucléoprotéine) dont une partie est complémentaire à l’unité de répétition télomérique.
Cet RNA agit comme un moule pour l’ajout de ces répétitions à l’extrémité 3’ par des cycles
répétés d’élongation et de translocation. Le brin complémentaire est synthétisé par le
mécanisme déjà décrit du brin retardé. L’amorce RNA à l’extrémité 5’ du brin riche en C est
- 27 -hydrolysée, si bien que le brin riche en G dépassera un peu par rapport au brin riche en C.
En plus, les télomères sont en fait une combinaison entre le DNA télomérique et des protéines
spécifiques constituant des structures appelées « capuchons télomériques »
- 28 -« CAPUCHONS
TELOMERIQUES
Dans les cellules somatiques et les cellules normales en culture, la télomérase est inactive (sa
production ou son activité est réprimée). Les télomères présents se raccourcissent, « s’usent »,
au fur et à mesure des réplications. Les chromosomes finissent donc par perdre leurs
télomères, ce qui les rend instables lorsque ce raccourcissement atteint le DNA codant : les
cellules deviennent sénescentes et meurent.
Par contre, dans les cellules germinales et dans les cellules cancéreuses, la télomérase est
active. Les cellules cancéreuses peuvent ainsi se multiplier indéfiniment sans que leurs
chromosomes se raccourcissent. C’est pourquoi, il existe plusieurs travaux de recherche
orientés vers la recherche d’inhibiteurs de la télomérase comme agents antitumoraux.
ζ- MODIFICATION POST-REPLICATION DU DNA.
Lors de la réplication, chez les eucaryotes, la cytosine est toujours incorporée non méthylée.
Après une première réplication d’un DNA méthylé sur ses deux brins, il apparaît donc un
DNA où un brin contient des cytosines méthylées tandis que l’autre brin contient des
cytosines non méthylées.
Il existe des enzymes appelées « maintenance méthylases ». Ces enzymes méthylent les
cytosines sur le nouveau brin de façon à maintenir l’information contenue dans la molécule
mère.
- 29 -II. MAINTIEN DE L’INTEGRITE DU DNA ET REPARATION
Toute cellule subit à tout moment des agressions physiques et chimiques de l’environnement
susceptibles d’altérer différentes molécules cibles. Les altérations de la molécule de DNA
seront les plus sérieuses puisqu’elles peuvent être pérennisées. Face à ces agressions, une
série de systèmes cellulaires assurent la maintien de l’intégrité du DNA et la réparation de la
majeure partie des altérations.
Les altérations se produisent principalement entre les mitoses et entraînent l’apparition de
mutations, de délétions et d’insertions. Les lésions responsables de ces altérations peuvent
correspondre à des dépurination, désamination, oxydation, etc.
Altérations possibles des
molécules de DNA
- 30 -Dans le cas des altérations provoquées par désamination, les bases modifiées obtenues
correspondent à des bases azotées inhabituelles au niveau du DNA (à l’exception du 5-
méthyl cytosine remplacé par de la thymine après désamination).
- 31 -Altérations d’origine physique :
Les « rayons cosmiques » et la « radioactivité », des rayonnements très énergétiques,
peuvent produire des lésions :
- Directes par modification des bases, rupture de brins, etc.
- Indirectes en provoquant l’apparition d’ « ions superoxydes » (O2-) chimiquement
très réactifs.
Les rayons ultraviolets solaires (UV), moins énergétiques, induiront principalement des
dimérisations de thymines adjacentes en créant un cycle cyclobutane entre les carbones 5
et 6 de chacune des thymines. Un bain de soleil d’une heure peut induire ainsi jusqu’à 80 000
dimères de thymines par cellule.
4
3 5
6
2
1
Altérations d’origine chimique :
Elles sont très diverses et peuvent même résulter simplement du métabolisme normal de la
cellule. Ainsi, les ions H+ cellulaires conjugués à l’agitation thermique peuvent retirer jusqu’à
104 bases puriques par jour et par cellule chez l’homme.
Effet des altérations :
Les altérations, quand elles ne sont pas immédiatement corrigées, peuvent entraîner des
modifications au cycle de réplication suivant.
- 32 -1. LE MAINTIEN DE L’INTEGRITE DU DNA EST ASSURE PAR DES
SYSTEMES DE SAUVEGARDE
α- FIDELITE DE LA REPLICATION
Nous l’avons déjà évoqué, la fidélité de la réplication n’est pas absolue. Nous avons vu que le
produit formé immédiatement après le passage de la polymérase comporte un taux d’erreurs
de 10-9. Malgré la propriété de correction d’épreuves des polymérases impliquées
« proofreading », certaines erreurs peuvent donc subsister.
Cependant, le taux d’erreurs réellement observé après réplication se situe en fait aux environs
de 10-11. Il existe par conséquent un système supplémentaire qui a pour fonction de :
- Détecter le mauvais appariement,
- Reconnaître le nouveau brin (et non pas le brin parental) où l’erreur a été commise et
le couper près de la mauvaise base pour procéder ensuite à la réparation.
Ce système, non encore complètement élucidé, semble faire intervenir une « recombinaison
post réplicative » entre brins fils et brins parentaux.
Chez les procaryotes, la discrimination entre le nouveau brin l’ancien repose sur le fait que
la méthylation du nouveau brin sur l’adénine des sites ‘GATC’ n’ont pas encore eu le temps
d’être effectués par la méthylase spécifique, alors que le brin qui a servi de matrice pour la
réplication est méthylé.
Chez les eucaryotes, où la méthylation s’effectue sur les cytosines des motifs ‘CG’, il est
vraisemblable que la reconnaissance du brin néosynthétisé utilise le même principe.
- 33 -β- SYSTEMES PREVENTIFS
Les cellules possèdent des systèmes de protection contre les agents susceptibles de provoquer
des altérations. Par exemple :
- Les ions superoxydes (02-) sont détruits par la « superoxyde dismutase ».
- Les ions H+ sont pris en charge par les systèmes de régulation de l’équilibre acido-
basique intracellulaire.
- Les oxydations sont réduites par différents systèmes plus ou moins spécifiques :
« NADPH2 », « glutathion », « vitamine E », etc.
En plus, chez les eucaryotes supérieurs, le rein et le foie assurent une fonction essentielle de
détoxification et d’élimination des substances chimiques néfastes.
2. LA REPARATION DU DNA EN DEHORS DE LA REPLICATION MET EN
JEU DES SYSTEMES MULTIPLES
α- REVERSION DIRECTE DU DOMMAGE
Les processus de réversions directes sont spécifiques de chaque type d’altération. Par
exemple :
- - Les « photolyases » sont des enzymes qui éliminent par photoréactivité les dimères
de thymine.
- Les guanines modifiées (6-O-méthyl-guanine) peuvent être déméthylées grâce à des
« 6-O-méthyl-guanine transférases ».
Remarque : La 6-O-méthyl-guanine s’apparie à la thymine dans le DNA
- 34 -β- REVERSION EN DEUX PHASES
PHASE 1 : Détection, suppression de l’altération
La première phase est la reconnaissance de l’altération. Chaque type d’altération possède un
système de reconnaissance qui lui est propre. Le résultat de cette phase est l’excision de la
base altérée ou d’une partie du brin de DNA où elle se trouve.
- Les glycosylases
L’action de ces enzymes (DNA glycosylases) va se
traduire par l’excision de la base azotée altérée
libérant un DNA ponctuellement dépuriné ou
dépyrimidiné. (Le même résultat peut être produit
directement par certains agents comme les ions H+).
Ensuite, il y a intervention des « AP endonucléases »
qui ont pour propriété d’effectuer une coupure
endonucléasique simple brin là où le DNA est
glycosylé.
REMARQUE : Dans le cas des 5-méthyl cytosines désaminées (→ thymine), il s’agit d’une
base ‘modifiée’ qui ne peut être reconnue par aucune des glycosylases ; de ce fait la
réparation ne sera jamais assurée. L’impuissance des systèmes de réparation dans ce cas
- 35 -précis fait que ce type de mutation a une fréquence apparente très supérieure à celle de la
plupart des autres mutations. Il s’agit de ce qu’on appelle un « point chaud de mutation » ou
« hot spot ».
- Système UVR
La réparation peut également faire appel au système « UVR A, B, C ». Un complexe
moléculaire reconnaît la lésion. Ce complexe est constitué de deux molécules de type A
« UVRA » associées à une molécule de type B « UVRB ». Sa fixation au DNA au niveau de
la lésion nécessite une molécule d’ATP. Le dimère de molécules A est libéré et il est
remplacé par une molécule de type C « UVRC ». Le complexe BC effectue une coupure
endonucléolytique de part et d’autre de la lésion puis se libère du DNA. Une hélicase se
charge alors de détacher le segment de DNA simple brin qui se trouve entre les deux
coupures. La brèche obtenue s’étend sur une trentaine de nucléotides.
PHASE 2 : Remplacement du DNA altéré
Cette phase consiste à remplacer les nucléotides manquant sous l’action conjuguée d’une
polymérase qui se sert du brin intact comme matrice et d’une ligase.
Chez les bactéries, la plupart des gènes codant pour les enzymes de réparation font partie d’un
système de sauvegarde complexe et hautement inter-régulé appelé le « système SOS ». Ce
système est composé d’une vingtaine de gènes dont les produits participent aux mécanismes
de réparation et de recombinaison. Deux gènes jouent un rôle central dans ce système : les
gènes recA et lexA.
- 36 -3. ANOMALIES DES SYSTEMES DE REPARATION CHEZ L’HOMME
Plusieurs maladies héréditaires ont été liées chez l’homme à un dysfonctionnement des
systèmes de réparation du DNA.
- 37 -Vous pouvez aussi lire

















































