DEMANDE DE CO-FINANCEMENT DE THÈSE DANS LE CADRE DU PROGRAMME AI_ENGINEERING_PHD@LILLE
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Demande de co-financement de thèse dans le cadre du
programme AI_Engineering_PhD@Lille
Intitulé du sujet de thèse : Développement de PROcédés CATalytiques d’Hydrogénation assisté par
Intelligence Artificielle
Acronyme du projet : PROCATHIA
Résumé de la thèse
L'objectif de la thèse PROCATHIA est de développer, grâce à des méthodes basées sur la chimie
théorique, l’intelligence artificielle et les expérimentations de criblage catalytique à haut débit, un nouveau
procédé catalytique d’hydrogénation d’anthraquinones alkylées. Cette réaction fait partie du procédé de
synthèse du peroxyde d’hydrogène (communément appelée eau oxygénée H2O2), un oxydant
respectueux de l’environnement, dont SOLVAY est le premier producteur à l’échelle mondiale. Ce procédé
mettra en œuvre des catalyseurs multimétalliques à base de palladium développés, soit par le partenaire
industriel SOLVAY lui-même, soit dans le cadre d’une autre thèse dirigée par le Prof. Dumeignil dans le
programme AI_PhD@Lille (thèse également cofinancée par SOLVAY).
Les principaux enjeux de la présente thèse PROCATHIA sont :
(i) de déterminer le mécanisme réactionnel de l’hydrogénation des anthraquinones alkylées sur le Pd
grâce à la DFT combinée aux outils de l’intelligence artificielle et à une validation expérimentale basée sur
les très nombreuses données issues de la plateforme REALCAT et des sites de production du partenaire
industriel SOLVAY ;
(ii) de développer des algorithmes de traitement des données issues des expérimentations de criblage à
haut débit et des sites industriels en s’appuyant sur l’intelligence artificielle afin de développer de véritables
outils d’aide à la décision pour les chercheurs et les industriels afin de préparer la prochaine génération
de procédé d’hydrogénation des anthraquinones alkylées.
Mots clés : catalyse, chimie industrielle, mécanisme réactionnel, DFT, algorithmes, intelligence
artificielle, peroxyde d’hydrogène, procédé, criblage haut-débit.
Contexte de la recherche
La thèse PROCATHIA sera réalisée au sein de l’Unité de Catalyse et Chimie du Solide (UMR CNRS8181)
sous la direction du Prof. Sébastien Paul, responsable de l’équipe VAALBIO et de la plateforme REALCAT.
La thèse sera co-encadrée par une ingénieure de recherche de SOLVAY localisée à Bruxelles : Dr. Jolien
De Waele et un chercheur CNRS en poste au laboratoire E2P2L (UMI CNRS/SOLVAY) localisé à
Shanghai, Chine : Dr. Jérémie Zaffran. Des chercheurs de CRIStAL, déjà impliqués dans le projet
REALCAT et la chaire industrielle SmartDigiCat, seront également en appui de ces travaux. A noter que
le co-financement de la thèse est déjà acquis chez SOLVAY. Notre partenaire industriel assurera 50% du
financement de la bourse de thèse ainsi que 17 k€ d’accompagnent par an (51 k€ au total sur 3 ans). En
outre il installera un dispositif expérimental d’une valeur de 15 k€ au sein de Centrale Lille dans les
laboratoires occupés par l’équipe VAALBIO de l’UCCS. Cette thèse s’inscrit dans le cadre du projet de
chaire industrielle SmartDigiCat porté par le Prof. Sébastien Paul (UCCS/VAALBIO/Centrale Lille) et
déposé à la MEL et à l’I-Site en décembre 2020 pour un financement mi-2021.
Directeur de thèse : Prof. Sébastien Paul (UCCS/Centrale Lille)
Co-encadrante SOLVAY : Dr. Jolien De Waele
Co-encadrant CNRS : Dr. Jérémie Zaffran
Présentation rapide des encadrants
Prof. Sébastien Paul, Professeur, 51 ans, sebastien.paul@centralelille.fr, +0033 320335457
Indicateurs bibliométriques1
H Index : 26 Nombre d’articles : 99
Nombre de citations : 2452 Nombre moyen de citations par article : 25
Sébastien PAUL est professeur à Centrale Lille, une école d'ingénieurs
généraliste du Nord de la France, où il a dirigé le département des sciences
de la matière entre 2009 et 2016. En 2019, il a été nommé directeur de
l'innovation et de la valorisation. Après son doctorat en génie chimique à
l'Université de Technologie de Compiègne, France (1996), il a été engagé par
Centrale Lille comme professeur assistant (1998). De 2004 à 2009, il a été à
la tête du département de génie chimique, qui est ensuite devenu le
département des sciences de la matière. En 2009, il a été nommé professeur
associé et, en 2011, professeur titulaire. Ses travaux de recherche sont menés
au sein de l'Unité de Catalyse et Chimie du Solide (UCCS - UMR CNRS 8181),
France, qui comprend plus de 300 personnes. Plus particulièrement, il dirige
le groupe VAALBIO (VAlorization of ALkanes and BIOmass). Sébastien PAUL
est également le coordinateur de la plateforme REALCAT ("Advanced High-
Throughput Technologies Platform for Biorefineries Catalysts Design") et le
responsable du laboratoire associé international franco-japonais du CNRS
NANOXCAT (2018-2022). Il a également fondé la start-up TEAMCAT SOLUTIONS en 2015. Il est impliqué
dans le développement d'une grande variété de procédés catalytiques à partir de composés issus de la
biomasse ou d'hydrocarbures dans le cadre de collaborations académiques et industrielles. Enfin, il a
supervisé ou co-supervisé 21 doctorats, 19 post-docs, est co-auteur de plus de 115 articles scientifiques
(hindex=26 - plus de 2400 citations), 18 brevets, et plus de 110 communications orales.
Dr. Ir. Jolien De Waele, Lab Application Manager & Chemistry Expert à Solvay, 29 ans,
jolien.dewaele@solvay.com, +32 2 264 17 32
Indicateurs bibliométriques
H Index : 3 Nombre d’articles : 3
Nombre de citations : 19 Nombre moyen de citations par article : 6
Jolien DE WAELE est Lab Application Manager & Chemistry Expert à Solvay,
une société avec ces activités dans des domaines matériaux et chimie. Elle a fait
une thèse sur le développement de catalyseurs bimétalliques pour la
déshydrogénation d’éthanol de 2014 à 2018. Pendant cette période, elle a publié
3 articles comme première auteur, a donné plusieurs communications orales et
a encadré 2 étudiants de master. En 2018, elle a été engagée par Solvay dans
la business unit Peroxydes comme Experte en Catalyse. Dans cette fonction, elle
est responsable de la recherche de nouveaux catalyseurs pour l’hydrogénation
des anthraquinones, qui fait partie du procédé de production d’eau oxygénée.
Elle a déjà encadré 2 stagiaires en catalyse. Fin 2020, elle est devenue manager
de l’équipe Applications & Stabilisants en plus des activités lié aux catalyseurs.
1 Data from www.scopus.com accessed the 26/06/2020
Dr. Jérémie Zaffran
J. Zaffran a obtenu son doctorat à l’École Normale Supérieure de Lyon (ENSL)
en 2014, sous la direction de Philippe Sautet dans le domaine de la
modélisation moléculaire de la catalyse hétérogène. Après un stage
postdoctoral au Technion-Israel Institute of Technology (IIT) en Israël, et un
séjour de recherche à l’Université ShanghaiTech en Chine, il rejoint en 2020
le laboratoire E2P2L en tant que chargé de recherche (CR) du CNRS. Son
champ d’expertise est lié au développement de modèles basés sur les
statistiques et l’intelligence artificielle, dans le but prédire de façon rapide et
systématique la réactivité des surfaces. Ce travail s’inscrit dans le cadre plus
global du « catalyst design » assisté par ordinateur, et permet d’accélérer
significativement la formulation de catalyseurs efficaces pour plusieurs types
de réactions. J. Zaffran est l’auteur d’une vingtaine de publications
scientifiques dans des journaux a haut facteur d’impact. Sa recherche trouve
des applications dans des domaines très variés, allant de la valorisation de la biomasse et du CO2 jusqu’à
l’élaboration d’électrodes photoactive pour les cellules photo-électrochimiques et les piles à combustible.
Présentation du projet, de la méthodologie, d’un ensemble de références
En tant que premier producteur et fournisseur mondial de solutions de peroxyde d'hydrogène (H2O2,
communément appelée eau oxygénée), Solvay dispose des qualités de ce composé les plus pures au
monde. Ce leadership a été obtenu grâce à une longue expertise, depuis 1951, et est maintenue grâce à
19 usines de production et 3 centres de recherche principaux répartis dans 15 pays à travers le monde.
Le peroxyde d'hydrogène est un agent oxydant couramment utilisé dans notre vie quotidienne, par
exemple pour le blanchiment du papier, les produits de lavage, l'exploitation minière et la production
d'oxyde de propylène qui est l'un des éléments de base de la production des mousses de polyuréthane.
Selon une étude de marché réalisée par Mordor Intelligence, le marché du peroxyde d'hydrogène pourrait
augmenter de 4 % en 20201.
Le peroxyde d'hydrogène est respectueux de l'environnement car il se décompose en H2O et O2 lors de
son usage. Sa production est réalisée à partir d'hydrogène et d'oxygène. Cependant, la mise en contact
directe de ces deux réactifs créerait un mélange explosif et est donc à éviter. Des recherches sont
néanmoins en cours pour tenter de maitriser cette technologie, mais le faible rendement obtenu et les
aspects très sensibles liés à la sécurité font que ce procédé n'est pas encore viable à l'échelle industrielle2.
Ainsi, pour éviter ce contact direct entre H2 et O2 la production de peroxyde d'hydrogène à l'échelle
commerciale est aujourd’hui très majoritairement réalisée par le procédé d'auto-oxydation (AO) 2 illustré
sur Figure 1. Dans ce procédé, une molécule d'anthraquinone alkylée (RQ) est hydrogénée en présence
d'un catalyseur à base de métal noble, généralement du palladium, et ensuite, la molécule est
spontanément oxydée en présence d'air avec formation simultanée de H2O2 et de la molécule
d'anthraquinone alkylée originale. Ce processus fonctionne donc en boucle, où la molécule
d'anthraquinone sert de molécule auxiliaire. La récupération de H2O2 à partir de la solution de travail, c'est-
à-dire la solution contenant la molécule d'anthraquinone hydrogénée (H2RQ) et ses solvants, s'effectue
par extraction. Une unité de réversion est également présente dans le processus pour transformer les
éventuels sous-produits en molécules utiles, c'est-à-dire en alkyl(tétra)quinone (hydrogénée). La version
"tétra" est la molécule originale d'alkyl anthraquinone dont l'un des cycles aromatiques latéraux est
hydrogéné (Figure 2). Les versions anthra et tétra peuvent toutes deux être hydrogénées et oxydées dans
le processus en boucle et sont donc désignées sous le nom de quinones utiles. Les autres sous-produits
sont des molécules d'(ox)anthrone et d'époxyde, produites respectivement dans les colonnes
d'hydrogénation et d'oxydation (Figure 2). Ces sous-produits peuvent être inversés dans l'unité de
réversion.
3/ EXTRACTION
H2O
+ H2O2
H2O2
+
2/ OXIDATION H2O
Auto-Oxidation
(AO)
Air
WORKING
SOLUTION
H2RQ
Side
Stream
(effluents)
H2
1/ HYDROGENATION 4/ REVERSION
Palladium catalyst
RQ
Figure 1: Représentation schématique de la synthèse de l’eau oxygénée par le procédé AO.
Figure 2: Représentation schématique des réactions principales dans les colonnes d’hydrogénation et d’oxydation du procédé AO.
Cependant, le Pd utilisé comme catalyseur pour la phase d’hydrogénation est considéré comme une
matière première critique par la Commission européenne3 [3]. Selon ses prévisions, l'approvisionnement
en palladium sera incertain d’ici 20 ans. De plus, un calcul approximatif a montré que, en considérant un
remplacement de 10 % de Pd par un autre métal dans le catalyseur employé par SOLVAY, le coût de
production du peroxyde d’hydrogène pourrait être réduit de 0,5 %, ce qui induit évidemment un flux de
trésorerie très important au regard des tonnages produits. Il y a donc un fort intérêt à réduire la quantité
de Pd utilisée pour synthétiser le catalyseur car une augmentation de son prix ou de sa consommation
pourrait rendre le procédé industriel moins rentable. En effet, depuis 2018, le prix du Pd a atteint des
sommets inégalés, ce qui rend son utilisation pour le catalyseur d’hydrogénation de plus en plus délicate
en termes de rentabilité économique du procédé. Dans le passé, le nickel de Raney était utilisé comme
catalyseur, mais le Pd a été choisi pour plusieurs raisons désavantageant le Ni : nécessité de travailler
dans des conditions alcalines, propriétés pyrophoriques et toxicité dans le produit peroxyde d'hydrogène,
même à faible concentration2. Cela indique cependant que l'hydrogénation de l'anthraquinone ne
nécessite pas nécessairement un catalyseur uniquement basé sur un métal noble.
De nombreux travaux sont en cours pour développer des catalyseurs bi- ou tri-métalliques qui sont souvent
supérieurs à leurs homologues monométalliques, en raison d'une synergie qui se produit lorsque plusieurs
métaux sont à proximité l'un de l'autre4,5,6. SOLVAY dispose déjà d’une bibliothèque de tels catalyseurs
qui seront utilisés dans cette présente thèse. Une autre thèse dirigée par le Prof. Dumeignil proposée
dans le cadre du programme AI_PhD@Lille (thèse également cofinancée par SOLVAY) visera également
au développement de ces catalyseurs.
Ici, un des enjeux majeurs de la thèse PROCATHIA sera de comprendre le mécanisme réactionnel de
l’étape d’hydrogénation sur Pd. Dans ce cadre, la DFT combinée aux outils de l’intelligence artificielle (IA)
avec validation expérimentale grâce aux nombreuses données issues de la plateforme REALCAT mais
aussi des sites de production du partenaire industriel SOLVAY apporteront une aide décisive.
Ponec et al.Erreur ! Signet non défini. ont rapporté que la sélectivité de l'hydrogénation des cétones-insaturées
est renforcée par la présence d'un mélange de sites métalliques et oxydés à la surface du catalyseur. Ceci
est lié au positionnement géométrique de la molécule à hydrogéner vis-à-vis de la surface du catalyseur
et donc des sites actifs. Ainsi, avec ce type de catalyseurs une meilleure sélectivité d'hydrogénation des
liaisons C=O par rapport aux liaisons C=C est obtenu. En effet, la molécule s'approche préférentiellement
de la surface de manière perpendiculaire. En outre, plusieurs auteurs, notamment Zhang et al. et
Mironenko et al.7 , 8, ont rapporté que le métal noble, dans le cas présent le Pd, joue un rôle à la fois
métallique et de site oxydé dans les catalyseurs monométalliques (Figure 3).
R R
H H H H
Pd0 Pd2+ Pd0 Pd0 Xy+ Pd0
Figure 3 : Représentation schématique de l’adsorption d’une molécule d’anthraquinone alkylée sur la surface d’un catalyseur
monométallique Pd (gauche) ou sur un catalyseur bimétallique à base de Pd (droite) [6].Dans un premier volet de cette thèse, nous étudierons le mécanisme réactionnel de l’étape
d’hydrogénation sur Pd en utilisant la théorie de la fonctionnelle de densité (DFT) combinée aux outils de
l’intelligence artificielle (IA) en association avec la validation expérimentale.
L’hydrogénation des anthraquinones alkylées RQ en RQH (Figure 2) se décompose en quatre étapes
élémentaires, auxquelles s’ajoutent l’adsorption de RQ et la désorption de RHQ de la surface métallique.
Le produit principal RHQ est en compétition avec de nombreux autres produits secondaires comme
représenté sur la Figure 4. Afin de connaitre la sélectivité d’une surface spécifique, il est nécessaire de
calculer l’énergie de chacun des états intermédiaires (IS) et des états de transition (TS) sur un chemin
réactionnel donné. Cette opération est particulièrement sensible car coûteuse en termes de temps de
calcul. C’est pourquoi afin de contourner les calculs DFT explicites, il est possible d’estimer l’énergie de
chacun de ces états avec une grande précision (par rapport à la DFT) en utilisant les outils de l’IA. Cette
stratégie, est basé sur l’approche dite « descripteur », populariser par Norskov dans les dernières
décennies.9 Elle consiste à prédire une énergie d’adsorption à partir d’une combinaison de descripteurs
pertinents comme le centre de la bande d d’un métal (εd), le nombre de coordination (CN) d’un élément,
la géométrie du centre actif (distances et angles de liaison), etc.
Dans un premier temps, on construira un échantillon d’apprentissage à partir de l’énergie d’adsorption de
tous les IS et TS menant à RHQ, ainsi qu’aux produits secondaires principaux. Ces énergies d’adsorption
seront calculées via DFT sur plusieurs facettes de références notamment la (111), la (100), la (121) et la
(110), déjà mentionnées pour leur efficacité.10 Plusieurs descripteurs de choix seront également calculés
en même temps pour chaque système. L’ensemble de ces résultats permettra de générer un modelé
prédictif de qualité et de l’entrainer jusqu’à obtenir une précision suffisamment élevée pour prédire des
énergies DFT avec des erreurs < 0.05 eV. Une fois testé par la méthode de « cross-validation », les
modèles élaborés serviront à générer de façon rapide et automatisée les diagrammes d’énergie
correspondant aux chemins réactionnels de la production de RHQ et d’autres produits secondaires sur
plusieurs autres facettes. La technique d’analyse micro-cinétique, sera déterminante à ce niveau pour
calculer la vitesse globale de production de chaque espèce. Ce résultat permettra à terme de discriminer
les surfaces de Pd en fonction de leur activité et de leur sélectivité.
Cette sélectivité pourra être validée expérimentalement en vérifiant la nature et la proportion du produit
principal par des méthodes de spectroscopie ou de chromatographie en phase liquide. Les mesures
expérimentales d’activité catalytique pourront être accomplies de façon massive et parallèle grâce à la
plateforme de criblage haut-débit REALCAT. Les abondantes données expérimentales ainsi générées
pourront également servir à élaborer un modèle prédictif d’IA dont les performances seront comparées au
précèdent modèle théorique.
Figure 4 : Diagramme d’énergie de l’hydrogénation de RQ vers RHQ (en noir) et un produit non désiré quelconque (en rouge). IS, état
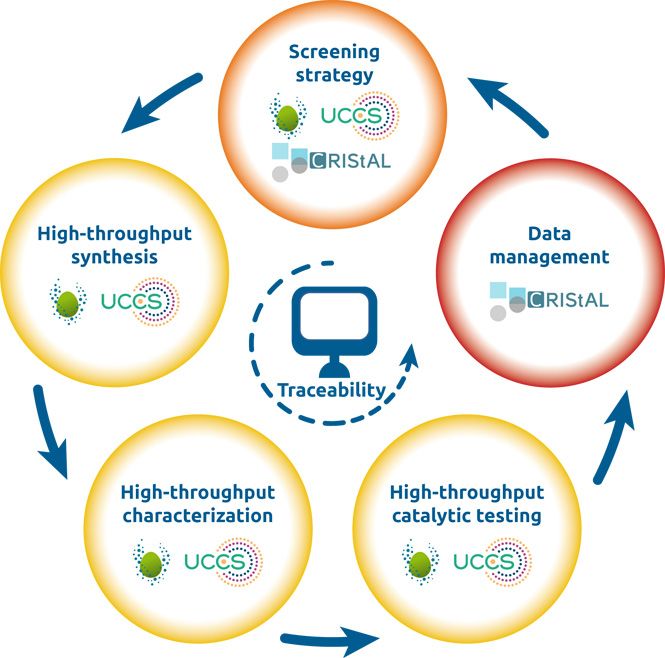
intermédiaire ; TS, état de transition.Dans un second volet de la thèse PROCATHIA, la force de frappe de la plateforme de criblage catalytique
haut-débit REALCAT (Equipex) disponible à l’UCCS sera employée afin de mettre en œuvre les
catalyseurs multimétalliques précités pour la réaction d’hydrogénation des RQ dans un très grand nombre
de conditions opératoires. Ces opérations robotisées, automatisées et parallélisées génèreront un volume
de données très important qui sera stocké dans la base de données de REALCAT (Figure 5). En outre, le
partenaire industriel dispose également d’un ensemble de données très important issues de l’exploitation
de ses sites de production de H2O2. Ils pourront venir enrichir cette base.
Stratégie
de criblage
1
Traitement
Synthèse
haut-débit
des
données
Apport de l’IA
2 5
Tracabilité
Caractérisation Test catalytique
haut-débit haut-débit
3 4
Figure 5 : Procédure itérative de développement d’un procédé catalytique en criblage haut-débit.
Un second objectif de la thèse PROCATHIA sera alors de développer des algorithmes de traitement de
ces données issues des expérimentations de criblage à haut débit et des sites industriels en s’appuyant
sur l’intelligence artificielle (IA) en utilisant comme cas d’application concret le développement de
catalyseurs pour la synthèse de l’eau oxygénée. L’idée est d’accélérer l’exploitation et l’interprétation de
ces données et de développer de véritables outils d’aide à la décision pour les chercheurs et les industriels
pour préparer la génération suivante de procédés catalytiques plus sûrs, plus respectueux de
l’environnement et plus efficaces. Ainsi, une approche d'apprentissage automatique (« machine learning »
noté ML dans la suite) capable d'établir rapidement la correspondance entre les données d'entrée du
système (c'est-à-dire les conditions de synthèse, les propriétés physico-chimiques ou encore les
conditions de tests des catalyseurs dans les réacteurs par exemple) et les données de sortie (rendement
en peroxyde d’hydrogène par exemple) sera mise en place. Une meilleure compréhension de la relation
entrée-sortie sera exploitée pour accélérer la découverte du bon catalyseur et de ses conditions de mise
en œuvre optimales grâce à la sélection rationnelle des conditions de synthèse et des conditions de
réactions les plus efficaces, en tirant le meilleur parti de la puissance de criblage haut-débit de la
plateforme REALCAT.
Le projet visera en outre à exploiter la richesse des données déjà existantes, qu’elles soient issues des
expériences précédentes ou à venir, notamment sur les sites de production de SOLVAY grâce à un
système de recherche intelligent. Le doctorant sera co-supervisé par des chercheurs expérimentés des
laboratoires de l'UCCS et de SOLVAY avec l’appui de ceux du laboratoire CRIStAL déjà très impliqués
dans la plateforme REALCAT et dans la chaire industrielle SmartDigiCat avec SOLVAY.
Dans le domaine de la catalyse, on ne trouve que des exemples très limités d'utilisation de l'intelligence
artificielle, en particulier lorsque des expériences catalytiques à haut débit et des techniques
d'apprentissage automatique ont été combinées pour accélérer les découvertes11,12,13,14. Le projet
PROCATHIA visera à aller plus loin en développant un cadre d'apprentissage automatique capable
d'établir rapidement les corrélations entre toutes les données d'entrée de différents formats (conditions de
synthèse ou propriétés physico-chimiques des catalyseurs, par exemple) et toutes les données de sortie
(rendement en molécules d'intérêt, par exemple) de la plate-forme REALCAT.La première étape de ce volet de la thèse consistera à consolider les données disponibles afin de les rendre fiables pour être traitées. Un catalyseur est d'abord synthétisé, puis caractérisé et testé pour la réaction ciblée (Figure 5). Par exemple, pour l'étape de synthèse, les données d'entrée comprennent les concentrations des différents réactifs, la température, le protocole utilisé, etc. Les données de sortie constituent la bibliothèque des catalyseurs. Nous devons accéder à toutes ces informations afin de pouvoir exploiter les similitudes entre les différents paramètres et protocoles pour construire des prédicteurs. La même approche sera répétée pour les étapes de caractérisation et de synthèse des catalyseurs. Notre base de données doit être robuste et continuellement enrichie au fil du temps. La deuxième partie du programme de recherche se concentrera sur le véritable défi que représente le traitement de l'ensemble des données. Un objectif important sera la reproductibilité de nos procédures de traitement afin que nous puissions garantir leur fiabilité. L'objectif final est de pouvoir prédire quelle formulation et quel protocole devraient être proposés pour synthétiser un catalyseur potentiellement efficace. Cet objectif a un avantage intermédiaire : il contribuera d'abord à optimiser notre plan expérimental d'exploration de l'espace des paramètres. Cela peut permettre de raccourcir la recherche d'une approximation de la région de l'espace des paramètres (formulation et protocole) qui peut conduire au catalyseur souhaité. Ensuite, en combinant des idées issues de la conception expérimentale et de l'apprentissage du renforcement, nous étudierons comment optimiser l'ensemble du processus. Deux tâches intermédiaires nécessiteront des recherches. La première tâche est la prédiction des propriétés physico-chimiques des catalyseurs connaissant leurs formulations et conditions de synthèse. La seconde tâche est la prédiction de l'efficacité d'un catalyseur lorsqu'il est utilisé dans la réaction chimique ciblée à partir de ses propriétés physico-chimiques. Notre objectif final pourrait résulter d'un couplage de ces deux prédicteurs, de sorte que nous serions alors en mesure de relier directement l'efficacité de réaction d'un catalyseur aux conditions de sa synthèse. Enfin, cela devrait conduire à la proposition d'un ensemble optimal de conditions de fonctionnement pour préparer les meilleurs catalyseurs avec les propriétés physico-chimiques optimales pour atteindre des performances élevées pour la réaction souhaitée. Aujourd'hui, toutes ces tâches d’exploitation des données sont effectuées manuellement, ce qui reste le goulot d'étranglement du processus de conception des catalyseurs : les résultats de cette thèse de doctorat pourraient rendre ce traitement de données automatisé, plus court et reproductible. Plus loin encore, on pourrait imaginer d'exploiter les idées issues de l'apprentissage par transfert pour exploiter les prédicteurs appris d'un milieu à l'autre, de sorte que chaque recherche sur les catalyseurs profite de toutes les autres. Ce sujet de thèse sera éminemment pluridisciplinaire, combinant à la fois la chimie théorique et expérimentale, l’intelligence artificielle et l’apprentissage automatique. Théoriciens et expérimentateurs fonctionneront en étroite collaboration et il y aura également un lien fort avec une seconde thèse dirigée par le Prof. Franck Dumeignil sur la synthèse des catalyseurs (proposée dans le cadre du programme AI_PhD@Lille - thèse également cofinancée par SOLVAY). En effet, une fois que par notre modèle théorique nous aurons identifié un set de facettes métalliques intéressantes pour leur activité et les sélectivités pour un produit donné, ces informations serviront à synthétiser des catalyseurs efficaces en exposant des facettes spécifiques. Cette opération peut être réalisée par le biais de promoteurs particuliers, comme déjà rapporté pour d’autres systèmes catalytiques dont certaines formes géométries bien définies ont pu être stabilisées avec Na et Mn.15
Indications sur le déroulement prévu de la thèse
L'organisation du doctorat est représentée de manière très schématique dans le diagramme de Gantt
suivant.
Temps (en mois) 1 6 12 18 24 30 36
1 Bibliographie – Analyse des données de réaction disponibles
2 Etude du mécanisme réactionnel (approche théorique)
3 Algorithme de traitement des données de réaction seules
(système simple)
4 Généralisation à un système complexe englobant les
données de synthèse, caractérisation et de réaction
5 Publication d’un premier article
6 Rédaction et soutenance de la thèse
Le doctorant bénéficiera de toutes les ressources des laboratoires de l'UCCS et de SOLVAY pour la
recherche bibliographique, les travaux expérimentaux et les calculs sur ordinateur. La principale difficulté
réside dans le caractère multidisciplinaire du sujet. En effet, il pourrait être difficile de trouver un candidat
au doctorat intéressé à la fois par la chimie (catalyse) et le traitement des données (intelligence artificielle
et apprentissage machine). Cependant, le directeur de thèse est fermement convaincu que la future
percée scientifique se fera à l'interface entre différentes disciplines et il utilisera cet argument pour
convaincre d'excellents candidats de leurs domaines de compétence de postuler au doctorat PROCAT_IA.
Il est à noter que le directeur de thèse dispose d'un très bon réseau international qu'il activera pour trouver
un excellent doctorant pour ce projet.
Motivations des encadrants indiquant en quoi leur sujet entre dans le cadre du programme
AI_Engineering_PhD@Lille
La thèse proposée ici envisage une utilisation de techniques d’apprentissage automatique et d’algorithmes
complexes d’intelligence artificielle pour le développement de procédés catalytiques importants pour
rendre l’industrie chimique plus sûre, plus respectueuse de l’environnement et plus efficace. Ce domaine
de recherche souffre de la grande complexité des phénomènes chimiques et physiques impliqués dans
les processus catalytiques ce qui rend leur prédiction impossible. Une approche expérimentale en « essai-
erreur » est encore nécessaire et le développement de procédés catalytiques industriels reste un
formidable défi qui requiert des temps de développement pouvant s’étendre parfois sur des dizaines
d’années. Ce travail, s’il est fructueux, aura un impact fort sur ce secteur de recherche car il propose une
approche pluridisciplinaire innovante dans le domaine de l’ingénierie des procédés catalytiques. Cette
approche sera ici appliquée à un procédé très important pour notre partenaire industriel SOLVAY : la
synthèse de l’eau oxygénée. Cependant, son champ d’application sera facilement étendu à d’autres
réactions impliquant des réactions d’hydrogénation. Les modèles prédictifs développés dans cette thèse
pourront également être enrichis afin d’être utilisables pour d’autres réactions que l’hydrogénation et
d’autres catalyseurs non-métalliques. La portée des travaux réalisés dans cette thèse s’en trouvera alors
largement agrandie. C’est pourquoi, nous sommes convaincus que ce projet a toute sa place dans le
programme AI_Engineering_PhD@Lille.Références bibliographiques 1 M. Intelligence, Hydrogen Peroxide Market - Growth, Trends, and Forecast (2020 - 2025), Mordor Intelligence, Research and markets, 2020. 2 G. Goor, J. Glenneberg, S. Jacobi, J. Dadabhoy, E. Candido, Hydrogen Peroxide, Ullmann's Encyclopedia of Industrial Chemistry, pp. 1-40. 3 D. Sustainability, B.G. Survey, B.d.R.G.e. Minières, N.O.f.A.S. Research, Study on the review of the list of Critical Raw Materials, in: European Commission (Ed.), 2017, pp. 515. 4 B. Coq, F. Figueras, Structure–activity relationships in catalysis by metals: some aspects of particle size, bimetallic and supports effects, Coordination Chemistry Reviews, 178-180 (1998) 1753-1783. 5 A.K. Singh, Q. Xu, Synergistic Catalysis over Bimetallic Alloy Nanoparticles, ChemCatChem, 5 (2013) 652-676. 6 H.-L. Jiang, Q. Xu, Recent progress in synergistic catalysis over heterometallic nanoparticles, Journal of Materials Chemistry, 21 (2011) 13705-13725. 7 J. Zhang, K. Gao, S. Wang, W. Li, Y. Han, Performance of bimetallic PdRu catalysts supported on gamma alumina for 2- ethylanthraquinone hydrogenation, RSC Advances, 7 (2017) 6447-6456. 8 R.M. Mironenko, O.B. Belskaya, T.I. Gulyaeva, M.V. Trenikhin, A.I. Nizovskii, A.V. Kalinkin, V.I. Bukhtiyarov, A.V. Lavrenov, V.A. Likholobov, Liquid-phase hydrogenation of benzaldehyde over Pd-Ru/C catalysts: Synergistic effect between supported metals, Catalysis Today, 279 (2017) 2-9. 9 J. K. Norskov, T. Bligaard, J. Rossmeisl, C. H. Christensen, Nat Chem 2009, 1, 37-46. 10 T. Kamachi, T. Ogata, E. Mori, K. Iura, N. Okuda, M. Nagata, K. Yoshizawa, The Journal of Physical Chemistry C 2015, 119, 8748-8754. 11 K. Takahashi, I. Miyazato, S. Nishimura and J. Ohyama, Unveiling Hidden Catalysts for the Oxidative Coupling of Methane based on Combining Machine Learning with Literature Data, ChemCatChem, 10, 3223 – 3228, 2018. 12 H. Wang, X. Chen, C. Li, Y. Liu, F. Yang, and C. Wang, Sequence-Based Prediction of Cysteine Reactivity Using Machine Learning, Biochemistry, 57, 451−460, 2018. 13 Z. Li, S. Wang, W. Shan Chin, L. E. Achenie and H. Xin, High-throughput screening of bimetallic catalysts enabled by machine learning, Journal of Materials Chemistry A, 5, 24131–24138, 2017. 14 Z. Li, X. Ma and H. Xin, Feature engineering of machine-learning chemisorption models for catalyst design, Catalysis Today, 280, 232–238, 2016. 15 Z. Li, T. Lin, F. Yu, Y. An, Y. Dai, S. Li, L. Zhong, H. Wang, P. Gao, Y. Sun, M. He, ACS Catalysis 2017, 7, 8023-8032.
Vous pouvez aussi lire

















































