Mélanie Eyries Florent Soubrier Département de Génétique Pitié-Salpêtrière APHP - FAVA-Multi
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Quel tableau clinique
quel tableau clinique justifie
justifie une une recherche
recherche de mutationsde
? mutations ?
quel tableau clinique justifie une recherche de mutations ?
Mélanie Eyries
Florent Soubrier
Département de Génétique
Pitié-Salpêtrière APHP
Le point de vue du laboratoire:
Justifié si recherche de mutation positive
MRO : les gènes responsables reconnus
quel tableau clinique justifie une recherche de mutations ?
Maladie génétiquement hétérogène
4 gènes connus appartenant à la voie de signalisation BMP
(2013) 85% mutations identifiées
(1994)
(2004) ~2% mutations identifiées
Taux de mutation en fonction du phénotype
78%
80%
70%
60%
% de mutations
50%
40% 18%
30%
20% 0%
10%
0%
≥3 2 1
Nombre de critères de Curaçao
68%
70%
55%
53%
60% 49%
50%
% de mutations
40%
30%
20%
10%
0%
Epistaxis Télangiectasies Manifestations Hérédité
Viscérales N=330
Types de critères de Curaçao
Taux de mutation identifiée dans MRO ≥3 ~85% :
les causes possibles
Exploration incomplète des gènes connus
- Mutations introniques profondes : effet sur épissage
- Mutations de régulation => hémizygotie transcriptionnelle :
promoteur, enhancer
MUTATION PROMOTEUR ENG
2 familles HHT sans mutation identifiée dans les gènes connus
ENG c.-127C>T dans les 2 familles
Famille A
Famille B
Décrit 2 fois dans la littérature : fréquence d’environ 3%
Dans 1 famille sur 3 testées négatives
Chez 4 individus sur 154 testés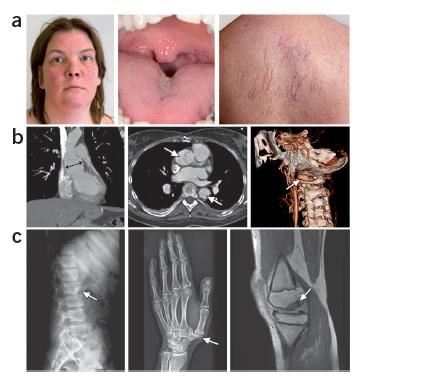
Taux de mutation identifiée dans MRO ≥3 ~85% :
les causes possibles
Exploration incomplète des gènes connus
- Mutations introniques profondes : effet sur épissage
- Mutations de régulation => hémizygotie transcriptionnelle :
promoteur, enhancer
Séquençage des gènes complets :
filtration des variants par coségrégation, cDNA, …
Phénotypes incomplets ou frontières => Nouveaux
gènes
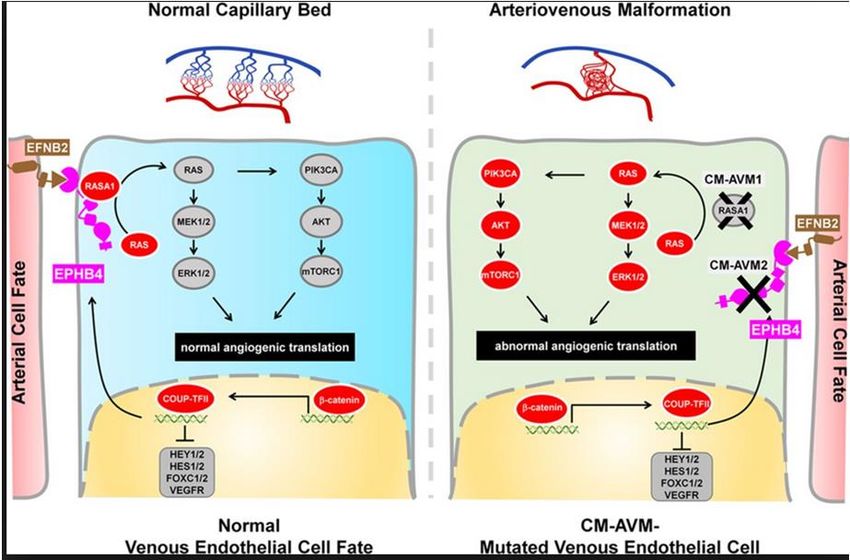
MUTATIONS RASA1 : CM-AVM1
+/-
Capture NGS 01/01/1950
Télangiectasies
Sanger MUT RASA1 +/- Sanger
ALK1/ENG/SMAD4 ALK1/ENG
RAS RAS
14/04/1974
02/04/1980 Epistaxis
Sanger Télangiectasies +/-
ALK1/ENG/SMAD4 Insuffisance veineuse
RAS
MAV cérébrale
Télangiectasies
Epistaxis
MUTATIONS EPHB4 : CM-AVM2
8 mutations identifiées sur 135 cas testés
Pathologie Malformations
Epistaxis MAV Hérédité
suspectée capillaires
1 CM-AVM Non Oui Non Oui
2 CM-AVM Non Oui Non Oui
Non
3 MRO (Possible) Oui Oui
explorées
Non
4 CM-AVM Non Oui Non Oui
5 CM-AVM Non Oui Non Oui
Oui (Veine
6 CM-AVM Non Oui
de galien)
Oui
7 MRO (Possible) Non Oui Non Oui
8 CM-AVM Non Oui Non Oui
UN PHENOTYPE A EXPLORER : LES FISTULES ARTERIO-VEINEUSES
CEREBROSPINALES DE L’ENFANT
Genetic Analysis in paediatric cerebro-spinal
arteriovenous malformation
• Point mutations and large rearrangements of ALK1, ENG and RASA1
genes
• A germline mutation was identified in 23/43 probands (53.5%±14.9%)
Genetic mutation None HHT RASA1 p value Test
9
Number of patients 20 ENG=8; 14
ALK1=1
Median follow-up (year) (IQR 1-3) 5 (2.5-10.5) 11 (8-17) 8 (5-15)
Mean age at onset (months) (SE) 21.6 (9) 60.8 (23.1) 22.8 (16.8) 0.20 Anova
Gender (M/F) 13/7 3/6 9/5 0.23 Chi2Mutation de novo Dans notre cohorte 15 mutations de novo identifiées sur 765 CIX mutés (114 pour lesquels les parents ont été testés ) Exemple de mosaïque germinale ACVRL1 :
Syndrome de Marfan et pathologies apparentées
Quel tableau clinique justifie une recherche de mutation?
N. HANNA – P. ARNAUD –C. BOILEAU
Département de Génétique
Hôpital Bichat
22 juin 2018 – 4ème Journée annuelle FAVA-MultiPré-requis à l’analyse moléculaire: Réglementaire: Consentement et attestation de consultation Fiche de renseignements cliniques CR écho cardiaque, examen ophtalmo si possible CR de consultation à faire suivre Bon de commande Suspicion MFS: atteinte de 2 systèmes min, dont au moins une atteinte majeure Toute demande particulière, ou demande de traiter le prélèvement « en urgence » doit être discutée avec le labo.
• Formes syndromiques
• Marfan syndrome (MFS)
• Loeys Dietz syndrome (LDS)
• Aneurysm Osteoarthritis
Syndrome (AOS)
• Beals syndrome
MFS LDS AOS
• Shprintzen-Goldberg syndrome
• …
• Formes non syndromiques
• Dilatation/Dissection Ao Ascendante (TAAD)
• Formes familiales prouvées
• Cas sporadiques < 45 ans sans FDR (bicuspidie, HTA…)
• Ectopie cristallinienne isoléeACTA2
NGS techniques ADAMTSL4
BGN
Successive Sanger COL3A1
sequencing EFEMP2
FBN1, TGFBR2, FBN1
FBN1 gene FBN2
TGFBR1, SMAD3,
FLNA
TGFB2, ACTA2, FBN2, Multiplicom FOXE3
ADAMTSL4 (Agilent) HCN4
Capture panel LOX
(MFS-TAA-V6, 28 gènes) LTBP2
LTBP3
Multiple genes MAT2A
panel MFAP5
MYH11
- TSCA (Illumina) MYLK
MLPA analysis for - Haloplex NOTCH1
FBN1 and TGFBR2 in (Agilent) PRKG1
MFS SKI
- Nimblegen
SLC2A10
(Roche) SMAD2
SMAD3
SMAD4
TGFB2
TGFB3
TGFBR1
TGFBR2NGS → Changement de stratégie moléculaire
Suspicion Marfan: TAAD Ectopie du Atteinte
1 signe majeur (familial et/ouPr Xavier Jeunemaitre – Dr Salma Adham – M Jean-Michaël Mazzella Centre de référence des maladies vasculaires rares, HEGP 4e journée annuelle FAVA-MULTI, 22 juin 2018 QUEL TABLEAU CLINIQUE JUSTIFIE UNE RECHERCHE DE MUTATION ?
Syndrome d’Ehlers Danlos vasculaire
– Freq estimée 1/150000
– Transmission AD – de novo 50%
– Gène COL3A1
• Expression
– Vaisseaux, peau, utérus et colon
– Signes mineurs
– Signes majeurs et complications
• Anévrysmes/dissections artériels
• Rupture colon sigmoïde Drera et al., 2011
• Age médian de survenue : 29 ans
Etat actuel de prise en charge ?
Progrès thérapeutiques ?Critères de Villefranche 1997 2 critères cliniques majeurs = proposer le test génétique
Critères révisés New York City 2017
Analyse des critères diagnostiques cliniques
(Henneton et al. Circ CV Genet, en revision)Analyse des critères diagnostiques cliniques
(Henneton et al. Circ CV Genet, en revision)Proposition de score de dépistage par test génétique dans le SEDv (Henneton et al. Circ CV Genet) : à valider de façon prospective
Vous pouvez aussi lire

















































