Implications potentielles de l'utilisation de l'information génétique numérique1 sur les ressources génétiques sur les trois objectifs de la ...
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Implications potentielles de l’utilisation de
l’information génétique numérique 1 sur les ressources
génétiques sur les trois objectifs de la Convention sur la
diversité biologique
Document soumis par le CGIAR au Secrétariat de la Convention sur la
diversité biologique i
Résumé
Le Secrétariat de la CDB a invité les organisations compétentes et les parties prenantes « à communiquer
des points de vue et des informations pertinentes sur l’utilisation de l’information génétique numérique
sur les ressources génétiques susceptible de concerner les trois objectifs de la Convention ». Le CGIAR
conduit une recherche stratégique visant le développement de l’agriculture pour assurer la sécurité
alimentaire. Sa mission a pour objet de bénéficier aux petits agriculteurs dans les pays en développement.
Les expériences du CGIAR acquises à ce jour confirment que les données numériques de séquences
génomiques2 peuvent jouer un rôle important dans la gestion et l’exploitation durable de la diversité
biologique et dans le partage des avantages découlant de l’utilisation de cette diversité. Pour ce qui est
de la préservation, des données numériques de séquences génomiques ont été utilisées pour évaluer la
diversité génétique de collections ex situ et pour identifier le germoplasme unique qui se trouve dans les
champs des agriculteurs et qui manque dans les collections ex situ ; cette information de base est
essentielle pour pouvoir élaborer des stratégies de préservation ex situ et in situ efficaces. Pour ce qui est
de l’exploitation durable, l’information sur les séquences génomiques, conjuguée aux données
phénotypiques et autres données peut être utilisée pour identifier des génotypes bien adaptés aux
différentes conditions agroécologiques changeantes. Intégrée dans les programmes de sélection végétale,
l’information sur les séquences génomiques s’avère de plus en plus utile dans l’obtention d’utilisations
plus rationnelles de la diversité génétique dans l’agriculture durable. L’avantage à partager le plus
important découlant de l’utilisation de l’information sur les séquences génomiques dans la recherche et
1
La terminologie doit faire l'objet d’un nouvel examen dans le cadre de l'étude et au sein du groupe d'experts.
2
Par « séquence génomique » on entend les séquences dérivées de l'ADN et de l'ARN. Elle inclue les lectures
courtes et longues et tous les marqueurs moléculaires dérivés tels que les polymorphismes à nucléotide simple
(SNP en anglais).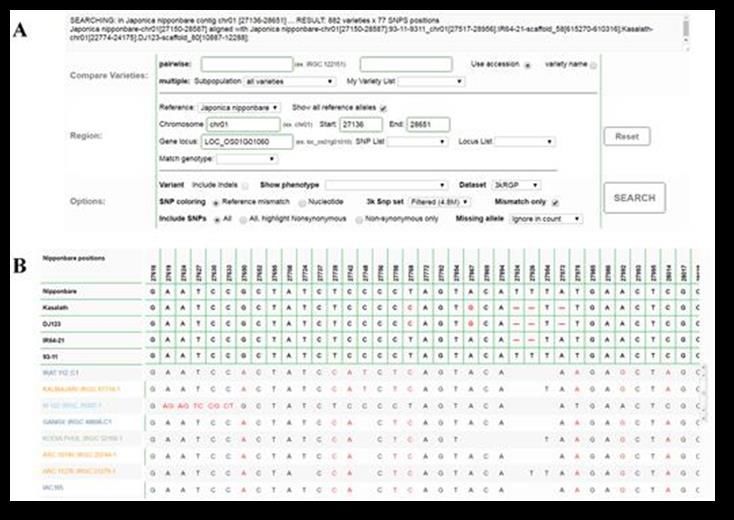
le développement agricole et la sélection végétale est qu’elle permet d’améliorer la sécurité alimentaire
et les moyens de subsistance. Parmi les autres avantages non monétaires résultant de l’exploitation de
l’information sur les séquences génomiques par les Centres du CGIAR, citons le meilleur accès des
agriculteurs aux technologies, le renforcement des capacités institutionnelles des organismes de
recherche des pays en développement, le partage des résultats de recherche ainsi que le développement
économique local et régional. Les avantages monétaires liés à l’exploitation des RPGAA par les Centres
sont en grande partie soumis au système multilatéral d´accès et de partage des avantages du Traité
international sur les ressources phytogénétiques pour l’alimentation et l’agriculture (TI-RPGAA). Le
système multilatéral réglemente l’accès aux ressources génétiques, mais pas celui aux informations sur
les séquences génétiques. Une des options actuellement à l’étude pour réviser le système multilatéral
consisterait à introduire un système de souscription. Cette souscription pourrait conduire à supprimer la
distinction entre l’accès et l’utilisation des ressources génétiques et l’information génomique, puisque le
partage des avantages reposerait sur le total des ventes de semences. Ceci permettrait de mettre en
évidence les avantages qui découlent de l’accès et de l’utilisation des ressources génétiques et de
l’information sur les séquences génomiques pour les utilisateurs commerciaux.
Les capacités technologiques de générer de l’information sur les séquences génomiques connues sous le
nom de technologies de séquençage de nouvelle ou de prochaine génération ont évolué plus rapidement
que celles qui permettent d’exploiter ces données. Comme les investissements dans la production initiale
de séquences génomiques sont relativement faibles, il importe d’investir plus dans l’analyse génomique
comparative, dans la connexion de la variabilité génétique à des traits phénotypiques utiles ou à la
performance, dans l’optimalisation de ces traits, et à terme dans la mise au point de nouvelles variétés
pour l’homologation et l’utilisation dans les champs des agriculteurs.
Les expériences acquises par le CGIAR dans la génération et l’utilisation des séquences génomiques sont
relativement récentes même si nous avons fait bien des progrès dans l’analyse des collections de
germoplasme. L’information sur les séquences génomiques devrait jouer un rôle accru dans les
programmes de préservation des ressources génétiques et de sélection menés par le CGIAR. Elle
permettra de créer des avantages pour les agriculteurs démunis de ressources dans les pays en
développement. Le CGIAR souligne l’importance du renforcement des capacités des organisations
chargées de RDI dans les pays en développement afin qu’elles puissent générer et utiliser l’information
sur les séquences génomiques dans le cadre de leurs propres programmes de préservation et
d’amélioration des espèces cultivées, et participer à des programmes de recherche et développement
coordonnés à l’échelle internationale et ce, sur un pied d’égalité. Le CGIAR a, entre autres, pour mission
de fournir à des partenaires nationaux dans les pays en développement les moyens de tirer avantage de
ces technologies, mais aussi d’autres technologies en pleine mutation, potentiellement révolutionnaires.
Pour ce faire, les Centres du CGIAR dispensent des formations et favorisent le transfert de technologies
aux scientifiques dans les pays en développement de manière à ce que les données de séquences
numériques puissent profiter à tous.
2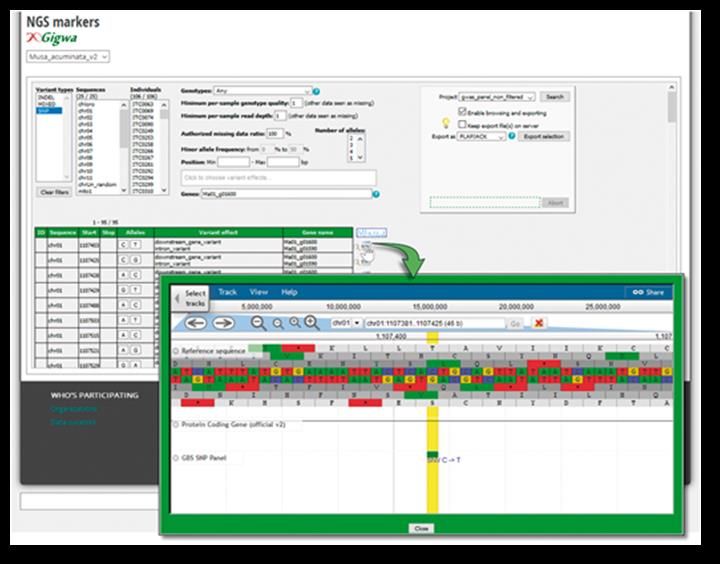
1. Introduction
Ce rapport est présenté par le CGIAR en réponse à l’invitation du Secrétariat de la CDB formulée à
l’encontre des organisations compétentes et des parties prenantes « à communiquer des points de vue
et des informations pertinentes sur l’information génétique numérique sur les ressources génétiques
susceptible de concerner les trois objectifs de la Convention ».
CGIAR
Le CGIAR est un partenariat mondial de recherche agricole qui s’emploie à assurer la sécurité alimentaire.
Les activités scientifiques du CGIAR sont axées sur la réduction de la pauvreté, l’amélioration de la sécurité
alimentaire et nutritionnelle, des ressources naturelles et des services écosystémiques. Menées au travers
de 15 centres, en étroite collaboration avec des centaines de partenaires, les activités de recherche du
CGIAR impliquent des instituts de recherche nationaux et régionaux, des organisations de la société civile,
des académies, des organisations de développement ainsi que le secteur privé. Notre mission consiste à
faire avancer les sciences et l’innovation agricoles de manière à donner aux pauvres et notamment aux
femmes la possibilité de mieux nourrir leur famille et d’améliorer leur productivité et leur résilience pour
que les personnes affectées puissent partager leur croissance économique et gérer les ressources
naturelles face au changement climatique et autres défis. Du point de vue géographique, la RDI menée
par le CGIAR se concentre sur les pays et les régions en développement.
Vu la nature de notre mission, de nos réseaux et de notre mode opératoire, les expériences que nous
avons acquises dans l’utilisation des données de séquences génétiques portent surtout sur la préservation
et l’exploitation durable de la diversité biologique agricole et le partage des avantages découlant de
l’utilisation des ressources génétiques pour l’alimentation et l’agriculture. Même si les Centres du CGIAR
ont quelques expériences dans la génération et l’utilisation de données de séquences génétiques des
animaux et des poissons, nous préférons nous concentrer dans le présent document sur les implications
de la génération et de l’utilisation des données de séquences découlant des ressources phytogénétiques
Objectifs de la Convention sur la diversité biologique
La CDB a trois objectifs tels que définis dans l’article premier :
(1) la conservation de la diversité biologique,
(2) l’utilisation durable de ses éléments, et
(3) le partage juste et équitable des avantages découlant de l’utilisation des ressources génétiques.
Après cette introduction, le reste du rapport se divise en trois sections séparées portant sur la contribution
actuelle et potentielle des données de séquences génétiques dans la réalisation de ces objectifs.
3
Diversité biologique agricole et ressources génétiques pour l’alimentation et
l’agriculture
Selon la Conférence des Parties à la Convention sur la diversité biologique (CDB) la biodiversité agricole
comprend « toutes les composantes de la diversité biologique constituant les (...) agro-écosystèmes : la
variété et la variabilité des animaux, des plantes et des micro-organismes, aux niveaux génétique, des
espèces et des écosystèmes, qui sont nécessaires au maintien des fonctions clés de l’agro-écosystème, à
sa structure et à ses processus ».3
La quasi-totalité des travaux menés par les Centres du CGIAR est axée sur la génération et l’utilisation de
données de séquences génétiques pour préserver et utiliser de manière durable la diversité génétique
intraspécifique et partager les avantages découlant de ces utilisations.
Qu’est-ce qu’on entend par « informations sur les séquences génétiques » (en
termes très généraux) et pourquoi celles-ci sont-elles utiles ?
Les constituants de base de vie de tout organisme vivant sur terre sont encodés dans son ADN. Les
séquences d’ADN sont uniques d’un organisme à un autre. Ces séquences peuvent contribuer à la
classification taxinomique, à l’identification des gènes uniques, à l’identification des combinaisons de
gènes qui encodent des traits précieux pour la production durable de denrées alimentaires dans un
environnement en évolution constante. Même si les séquences pangénomiques sont de plus en plus
accessibles, l’utilisation de ces informations pour créer des avantages est toujours bien en deçà de la
technologie permettant de générer des données de séquences. Actuellement, des marqueurs génétiques,
de minuscules segments d’ADN dispersés à travers le génome sont déployés avec succès pour génotyper
les individus et pour identifier avec un certain succès les individus dans les programmes de sélection qui
contiennent des traits d’importance en termes d’agronomie. Dans les programmes de préservation des
ressources génétiques, le génotypage est un outil fort utile pour déterminer les lacunes des collections et,
à l’avenir, pour assurer la préservation ainsi qu’une utilisation plus rationnelle d’une précieuse diversité
génétique unique. Pour les programmes liés à la conservation in situ, mais aussi pour les communautés
autochtones, les empreintes génétiques uniques des variétés de culture existantes peuvent être utilisées
pour établir des données de base pour les travaux à venir visant la préservation ou la prévention de la
perte des variétés détenues par des agriculteurs ainsi que pour documenter l’impact de la diversité dans
les champs des agriculteurs avec différentes méthodologies d’intervention y compris la réintroduction de
variétés natives conservées ex situ ayant disparu à l’échelon local.
Les variétés (variétés des agriculteurs, variétés sélectionnées ou ancêtres sauvages) représentent des
combinaisons de séquences génétiques qui étayent les traits de chaque variété particulière en interaction
3
Conférence des Parties à la Convention sur la diversité biologique. Décision V/5 Annexe : Diversité
biologique agricole : examen de la première phase du programme de travail et adoption d'un
programme de travail pluriannuel. CDB : Montréal, 2000.
4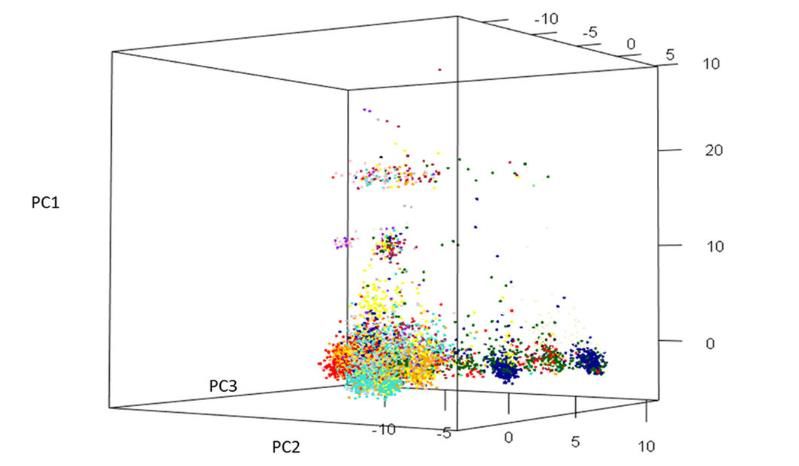
avec l’environnement dans lequel la variété est cultivée. Même si une variété est unique en son genre, les
séquences génétiques individuelles (p. ex. codification de variété précoce ou tardive) peuvent être
exprimées de la même manière dans beaucoup de variétés différentes. Le contraire peut également se
produire : le même trait, p. ex. la précocité peut également être sous le contrôle de différentes séquences
dans différentes variétés. L’expression de chaque gène donné peut être largement influencée par
l’environnement. La spécificité d’une variété provient de la combinaison de ces séquences génétiques et
de l’influence de l’environnement. Chez une variété particulière, la combinaison résulte de centaines voire
de milliers d’années de sélection aléatoire, environnementale, d’une sélection pratiquée par des
agriculteurs ou des obtenteurs.
L’information numérique génétique permet :
- aux gestionnaires de banques de gènes d’évaluer et de quantifier à sa juste mesure le degré de
variabilité (polymorphisme) parmi les individus d’un paquet de semences ou entre plusieurs
accessions de banque de gènes. Cela peut assurer le contrôle de qualité ainsi que la bonne
maintenance, la distribution et l’utilisation des banques phytogénétiques ;
- aux scientifiques spécialisés dans les espèces cultivées d’obtenir l’information génétique de base
requise pour pouvoir comparer les génotypes ex situ avec la diversité maintenue par les
agriculteurs dans des conditions in situ, pour améliorer l’appréhension de la diversité présente
dans ces espèces cultivées et ces animaux domestiqués et pour faire l’inventaire de ce qui existe
et de ce qui manque dans les banques de gènes ou dans les paysages communautaires ;
- aux sélectionneurs de saisir et d’utiliser la diversité qui existe pour pouvoir développer des
stratégies de sélection plus efficaces et atteindre les objectifs en termes de gain génétique ;
- aux pays membres, aux communautés locales et aux peuples autochtones de mieux quantifier et
de procéder à l’analyse d’empreinte de la diversité qu’ils détiennent in situ, en identifiant la
diversité qui est la plus menacée ainsi que la diversité qui est unique ou qui est utilisée par la
communauté ou dans les champs des agriculteurs ;
- aux gouvernements d’élaborer, en partenariat avec les agriculteurs et autres gestionnaires de
ressources naturelles (hommes et femmes confondus) des priorités en matière de préservation
pour maintenir la diversité, suivre les populations in situ au niveau génétique ainsi que pour
comprendre comment les populations répondent aux changements de température, de l’eau, des
engrais, des nutriments, du mode de gestion, etc.
Même si les données de séquences génétiques sont un outil puissant, elles ne sont pas une panacée et
elles ne peuvent pas non plus être utilisées de manière isolée. Il convient de les combiner avec d’autres
technologies. Certes, l’obtention de grandes quantités de données devient de moins en moins chère, mais
il n’y a pas suffisamment d’outils pour rassembler ces données sous une forme globale et utilisable pour
5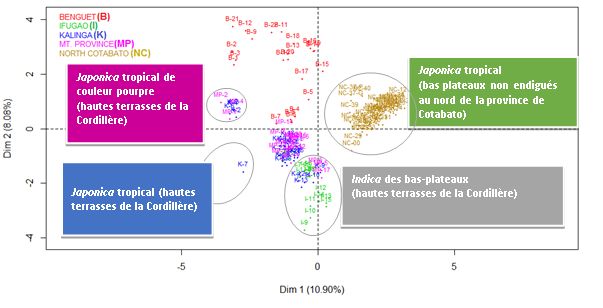
renforcer le gain génétique4 chez la plupart des espèces cultivées, le bétail et les poissons, ni dans
l’hémisphère nord ni dans l’hémisphère sud. En tant que leaders et membres de consortiums plus larges,
les Centres du CGIAR sont impliqués à des niveaux différents dans le séquençage pangénomique ou le
séquençage à haute densité et le génotypage d’espèces cultivées (y compris la banane, le manioc, le pois
chiche, le pois de vache, l’arachide, le millet, le maïs, le pois cajan, la pomme de terre, le riz, le sorgho, la
patate douce, le blé et l’igname).
Même si le séquençage du génome et l’empreinte génétique permettent de faire la distinction entre « ce
qui est identique » et « ce qui diffère » du point de vue génétique, le développement de ces outils est
entravé par le fait qu’il y a un manque de données (de phénotypage) morphologiques complémentaires.
Or celles-ci sont nécessaires pour pouvoir interpréter pleinement les données de séquences numériques.
La plupart des traits sont soumis à un contrôle génétique complexe comprenant de multiples formes de
familles de gènes en interaction en réseau. Par conséquent, il est difficile de comprendre un scénario de
cause à effet pour un gène donné par rapport à un trait donné. Par exemple, la tolérance à la sécheresse
d’une espèce cultivée dépendra de l’anatomie et de l’architecture des racines, des feuilles et des tiges de
cette espèce, mais aussi de l’avancement de la plante dans son cycle de vie par rapport au développement
de la sécheresse, ainsi que de certaines qualités difficiles à mesurer telles que la capacité
photosynthétique, la capacité respiratoire et les autres capacités biochimiques et physiologiques de la
plante (p. ex. la chute des feuilles). Aucun de ces mécanismes liés à la tolérance à la sécheresse n’est
simple et il n’y a pas de gène particulier pour répondre à la sécheresse. La situation est la même pour les
animaux : les traits clefs du bétail tels que le poids au sevrage, la tolérance à la chaleur et aux maladies
dépendent de multiples caractéristiques régulées par des myriades de gènes. Les Centres du CGIAR et ses
partenaires acquièrent actuellement de l’expérience dans l’usage du phénotypage à haut débit.
Cependant, l’opération est onéreuse et requiert des investissements importants en expertise et en
ressources.
En résumé, on peut dire que les données de séquences d’ADN en elles-mêmes sont de valeur restreinte,
même si leur valeur incrémentale est considérable. Les outils et méthodes actuellement développés
devraient permettre une meilleure exploitation de ces données. Les séquences pangénomiques peuvent
fournir des informations précieuses. Cependant, les marqueurs d’ADN sont présentement beaucoup plus
utilisés pour le génotypage et les tests visant l’appréhension de traits de base génétiques. Ces marqueurs
permettent d’accélérer la sélection et les gains génétiques et ce, à l’échelle du globe. Dans les pays en
4
Par « gain génétique » on entend l'augmentation de la performance de traits d'intérêt d'individus
sélectionnés soumis aux programmes d'amélioration génétique, entre la première et la deuxième
génération originale, lorsqu'elles sont comparées dans le même environnement.
6développement, beaucoup de programmes de sélection ont recours à ces approches pour permettre une
sélection rapide des variétés ainsi que des sélections avec des traits importants pour elles.
2. Contribution des informations sur les séquences génétiques dans
la préservation de la diversité biologique
Pour ce qui est de la préservation, les principales obligations des parties contractantes sont définies dans
les articles 8 de la CDB (« conservation in situ »), 9 (« conservation ex situ ») et 7 (« identification et
surveillance en liaison avec la conservation et l’utilisation durable »).5
En 2010, la Conférence des Parties à la CDB a adopté les objectifs Aichi. L’objectif 13 stipule ce qui suit :
« D’ici à 2020, la diversité génétique des plantes cultivées, des animaux d’élevage et domestiques et des
parents pauvres, y compris celle d’autres espèces qui ont une valeur socio-économique ou culturelle, est
préservée, et des stratégies sont élaborées et mises en œuvre pour réduire au minimum l’érosion
génétique et sauvegarder leur diversité génétique ».
Un des défis liés à la réalisation de l’objectif 13 est l’identification et la quantification de la diversité
génétique qui requiert une maintenance. Pendant longtemps, les gestionnaires des collections ex situ ont
essayé de définir la diversité des espèces cultivées qu’ils détiennent de manière à identifier et à préserver
les lacunes importantes (diversité manquante) dans les collections. La caractérisation de la diversité
trouvée in situ, et pour les espèces cultivées et pour le bétail, est également considérée comme l’un des
principaux objectifs poursuivis par les différents acteurs impliqués dans la préservation de la diversité
agricole. La caractérisation reste un objectif difficile à atteindre malgré le travail qui a été accompli dans
l’élaboration de méthodes de séquençage de l’ADN. L’objectif pourra néanmoins être atteint lorsqu’il sera
possible de séquencer des collections ex situ entières et des échantillons représentatifs de la diversité in
situ. Dans le passé, les descripteurs morphologiques ont été utilisés pour caractériser les collections ex
situ et distinguer les différences génétiques d’une adhésion à l’autre. Ce système fonctionne et il est
toujours considéré comme le moyen privilégié pour classer les collections ex situ. La même approche est
appliquée à l’étude des espèces cultivées et des populations animales in situ. Toutefois, ces marqueurs
ont montré leurs limites lorsqu’il s’agit d’identifier des variétés certes étroitement apparentées, mais
génétiquement distinctes et de définir l’étendue de la diversité génétique qui existe au sein des races
primitives de certaines espèces cultivées ou d’animaux domestiques, mais aussi entre elles. La
caractérisation de la diversité génétique à l’aide de descripteurs morphologiques risque d’être faussée à
cause de l’étendue de la plasticité environnementale de ces caractères morphologiques. Pour caractériser
la diversité et analyser l’ampleur de la diversité in situ, les données de séquences d’ADN constituent les
meilleurs outils disponibles, à condition de pouvoir accéder à des volumes suffisamment grands. Elles
5
L'annexe 1 fournit une liste des composantes de diversité biologique qui devraient être suivies, au
niveau de l'habitat, des espèces et du point de vue génétique. Le paragraphe 3 stipule : « Génomes et
gènes décrits revêtant une importance sociale, scientifique ou économique ».
7contribuent également à la préservation à long terme d’un maximum de diversité de sorte que tous les
éléments de la diversité présents dans chaque espèce végétale ou animale peuvent être préservés à long
terme pour l’humanité. Ainsi, les données de séquences d’ADN sont un moyen puissant et parfois c’est
d’ailleurs le seul outil qui permet aux gestionnaires de banques de gènes, aux organisations agricoles
nationales, aux entreprises, aux communautés autochtones et aux chercheurs d’atteindre pleinement cet
objectif d’Aichi et de quantifier la diversité présente, in situ et ex situ, pour ce qui est des principales
espèces végétales et animales domestiquées.
Depuis sa création, le CGIAR a investi dans la préservation de la diversité biologique agricole ex situ et
dans l’exploitation durable de cette ressource pour remplir sa mission. Le manque de connaissance de la
diversité génétique constitue un goulot d’étranglement et c’est la raison pour laquelle le CGIAR s’est mis
au pas des nouvelles technologies pour pouvoir mieux caractériser la diversité des espèces cultivées,
comprendre les liens entre les entrées conservées, identifier et combler les lacunes que présentent les
collections ex situ détenues par ses centres à l’échelle du globe pour la communauté internationale.
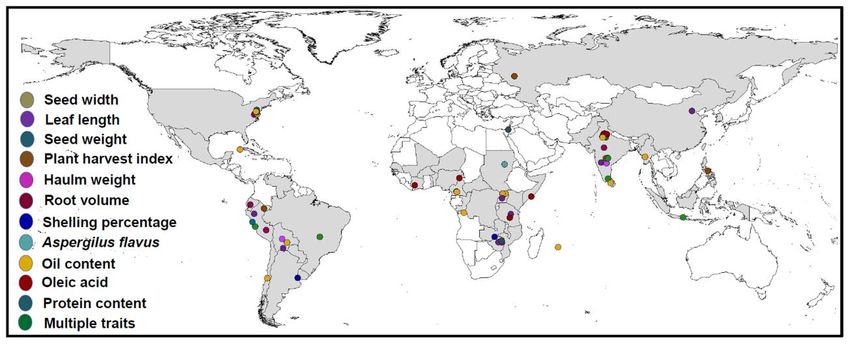
L’encadré 1 présente un exemple de l’expérience du CIP dans la caractérisation génétique d’un ensemble
de races primitives de pomme de terre issu de la génothèque ex situ. Des études comme celle-ci mettent
en évidence les liens de parenté des différentes espèces au sein des collections ex situ et peuvent être
utilisées pour l’identification d’allèles novateurs qui ne sont pas conservés ex situ lorsque le génotypage
similaire est exécuté in situ. L’encadré 2 présente la structure génétique des accessions détenues par l’IITA
dans sa collection ex situ de maniocs.
Encadré 1 : Accessions de génotypes du CIP pour comprendre la structure génétique de la collection ex
situ de patates douces
Traditionnellement, les agriculteurs péruviens cultivaient entre 20 et 40 races primitives de pommes de
terre. Cette diversité leur permettait de faire face aux éventualités. En effet, certaines races primitives
produisent une plante même durant les années difficiles, ce qui permet aux agriculteurs de survivre
jusqu’à la prochaine récolte. Ces dernières décennies, certaines communautés ont réduit
progressivement la culture de nombreuses variétés de pommes de terre, de sorte que beaucoup de
familles ne plantent plus qu’une dizaine de variétés. Le CIP mène un programme pour que ces variétés de
races primitives qui sont collectées dans ces régions soient restituées ou rapatriées de manière à
constituer des systèmes de sauvegarde pour que les communautés autochtones puissent poursuivre
leurs pratiques traditionnelles en matière de culture là où la diversité joue un rôle crucial dans la
durabilité à long terme. Le défi lié à ce travail de restauration est que nous ignorons l’étendue qu’avait
cette diversité il y a 50 ou 100 ans voire même la diversité qui existe actuellement dans la génothèque ex
situ. En conséquence, la génothèque du CIP a récemment procédé au génotypage de l’ensemble de sa
collection de races primitives de pomme de terre et de patate douce, posant ainsi le fondement de
l’évaluation de l’actuelle diversité dans la collection ex situ.
8Figure 1 : Dendrogramme de l’ensemble de référence de base de la diversité des variétés primitives de
pommes de terre détenues par le CIP (papa nativa) reposant sur les données génétiques issues de la
matrice SNP SolCAP 12k. Les lignes colorées dans le dendrogramme dénotent des espèces de pommes de
terre différentes (sur la base de Hawkes 1990) qui montrent un groupement clair par espèces. La barre
au-dessous du dendrogramme dénote le degré de ploïdie. Les données comme celles-ci permettent de
définir la diversité dans la collection et d’identifier les accessions individuelles d’intérêt pour une étude
plus détaillée. (Données inédites du CIP).
Encadré 2 : Appréhension de la structure et de la diversité génétique de la collection internationale de
maniocs détenue par l’IITA
9Analyse de cladogrammes de 340 échantillons issus de 319 accessions de manioc (25 sauvages, 281 races
primitives de diverses collections de base ex situ / de champ de l’IITA et 14 cultivées), génotypés à l’aide
de marqueurs SNP 38K DArTseq montrant la diversité génétique disponible et la relation entre les
accessions ayant des origines de différentes régions de l’Afrique occidentale (IITA, inédite). Les
différentes couleurs indiquent les pays d’origine.
Les Centres du CGIAR ont pour mission de maximaliser l’exploitation de leurs collections en caractérisant
les ressources phytogénétiques et en partageant les informations relatives au germoplasme qu’ils
détiennent. Cependant, la simple identification des traits pouvant susciter un intérêt ne suffit pas, car
l’étendue des collections rend pratiquement impossibles l’incorporation et l’exploitation de ces traits pour
améliorer les programmes. La création de sous-ensembles plus petits ou de sous-ensembles de référence
de base permet aux programmes de sélection de recourir à de tels traits, comme c’est le cas avec les races
primitives de blé mexicain présentées dans l’encadré 3, les collections ICRISAT dans l’encadré 4 et le riz
africain dans l’encadré 5. L’information génétique permet de créer judicieusement ces ensembles de base
pour assurer la représentation de la diversité des collections dans l’ensemble de base, et pour fournir aux
sélectionneurs des conseils quant à l’utilisation de ces ensembles de manière plus efficace.
Encadré 3 : Déverrouillage de la diversité génétique des variétés de blé créole
(résumé d’un document présenté par Vikram et al. 2016)
Ces races primitives de blé mexicain connues également sous le nom de « blés créoles » ont été
introduites aux Amériques du 16e au 18e siècle. Elles se sont progressivement adaptées aux conditions
locales environnementales, y compris dans de nombreuses régions soumises à la chaleur et à la
sécheresse. À ce titre, elles devraient comporter des variations génétiques utiles pour la tolérance au
stress. L’introduction de la diversité génétique de ces blés créoles dans les pipelines de sélection
pourrait aboutir à la mise au point de la prochaine génération de variétés de blé. Avec cet objectif à
l’esprit, une équipe de scientifiques du CIMMYT, l’Instituto Nacional de Investigaciones Forestales,
Agrícolas y Pecuarias (INIFAP) au Mexique et le Conseil de la biodiversité du Punjab (Inde) ont réalisé
une étude pour : (1) caractériser la collection de variétés de blé mexicain conservées dans la Banque de
matériel génétique CIMMYT ; et (2) développer un ensemble de référence de base défini à l’aide de
plusieurs variables. Un ensemble de référence de base est un sous-ensemble d’une collection de
ressources génétiques qui représente l’ensemble de la diversité de la collection, mais qui est
suffisamment restreint pour que les éleveurs puissent faire l’évaluation des traits intéressants.
Autrefois, les ensembles de référence de base étaient établis à partir d’une seule variable, par exemple
des données génotypiques, des mesures de phénotype ou une répartition géographique. L’utilisation
simultanée de plusieurs types de variables (génotype, phénotype, géographie, etc.) permet d’obtenir
une estimation fiable de la diversité susceptible de faire l’objet d’une application phytogénétique.
Résultat : 8 416 races primitives de blé représentant tout un éventail d’agroécologies mexicaines ont été
caractérisées à l’aide de marqueurs génétiques (DarTseq) et ont également fait l’objet d’un phénotypage
pour déterminer le potentiel de rendement, la tolérance à la sécheresse, à la chaleur, la résistance à la
rouille jaune, afin d’identifier un ensemble de référence de base susceptible de représenter cette
variation importante. Cet ensemble de référence de base regroupe 89 % des allèles rares présents dans
l’ensemble complet.
Figure 2 : présentée sous forme de graphique en 3D, l’analyse en composantes principales (ACP) montre
la répartition des groupes de races primitives du blé mexicain sur la base des marqueurs génétiques.
10Au total, on compte 15 groupes qui correspondent à différents États mexicains. 1 = jaune (MEXICO,
PUEBLA), 2 = bleu clair (MEXICO, QUERETARO), 3 = bleu foncé (CHIHUAHUA, OAXACA), 4 = orange
(MEXICO, PUEBLA, QUERETARO, HIDALGO), 5 = vert clair (DURANGO), 6 = vert foncé (CHIHUAHUA 95.5),
7 = rose (OAXACA, TLAXCALA, TOLUCA, PUEBLA), 8 = violet (OAXACA), 9 = turquoise (MEXICO),
10 = marron (MEXICO, MICHOACAN), 11 = rouge (COAHUILA), 12 = gris (TLAXCALA, MEXICO,
MICHOACAN), 13 = châtaigne (MICHOACAN), 14 = beige (CHIHUAHUA 95.5), 15 = noir (GUANAJUATO).
Les contributions de PC1, PC2 et PC3 s’élèvent respectivement à 10,5, 8,2 et 6,9 % de la variation totale.
Encadré 4 : Création de mini-collections de base pour exploiter davantage le germoplasme pour
l’amélioration des espèces cultivées
La diversité du germoplasme est à la base des programmes d’amélioration des espèces cultivées.
Cependant une grande partie du germoplasme détenu dans les génothèques n’a pas encore été utilisée
dans les programmes d’amélioration des cultures. Il est donc nécessaire d’utiliser plus de germoplasme
dans les programmes d’amélioration des espèces cultivées afin d’assurer une production agricole
durable et accrue pour la sécurité alimentaire. La génothèque ICRISAT conserve plus de
125 000 accessions de six plantes sous mandat et de cinq petits mils provenant de 144 pays. Cette
utilisation peu élevée du germoplasme s’explique par le manque d’information quant aux traits
d’importance économique et l’ampleur des collections. Pour améliorer l’utilisation du germoplasme
dans les programmes d’amélioration des espèces cultivées, des collections de base représentatives
(10 % de l’ensemble de la collection) ont été mises au point en utilisant des données quantitatives et
qualitatives sur les traits du pois chiche, du pois cajan, de l’arachide, du sorgho, du millet perlé, du mil
rouge, du millet des oiseaux, du millet commun, du pied-de-coq cultivé, de l’herbe à épée, de la miliade.
Malheureusement, le nombre d’accessions à ces ensembles de référence de base est toujours trop
important pour permettre une évaluation pertinente dans les programmes de sélection. Pour surmonter
ce problème, les scientifiques de l’ICRISAT (Upadhyaya et Ortiz, 2001) ont développé le concept de mini-
collection de base (10 % de la collection de base ou 1 % de l’entière collection) et ont proposé une
stratégie en deux phases en utilisant les données relatives aux traits de la mini-collection de base. Une
vaste évaluation multidisciplinaire de mini-collections de base a permis d’identifier de nouvelles sources
11de variation pour de multiples caractères y compris la tolérance aux stress biotiques et abiotiques et les
caractéristiques nutritionnelles et agronomiques. Ces mini-ensembles de référence de base ont été
distribués aux sélectionneurs dans 36 pays pour être utilisés dans des programmes d’amélioration. Le
séquençage de ces mini-collections de base serait d’une grande valeur pour déterminer les variations de
séquence associées aux traits et permettrait d’identifier les lignées de germoplasme les plus utiles pour
être utilisées comme parents dans les programmes de sélection.
Encadré 5 : Variation génétique et structure des populations d’Oryza glaberrima et mise au point
d’un ensemble de référence de base à l’aide d’un séquençage DArT
La génothèque AfricaRice détient deux espèces cultivées (Oryza sativa et O. glaberrima) et cinq
espèces sauvages (O. longistaminata, O. barthii, O. punctata, O. brachyantha et O. eichingeri), la
plupart étant d’origine africaine. Ces accessions de riz présentent des mécanismes d’adaptation ou de
protection aux différents stress abiotiques et biotiques, mais sont généralement caractérisées par un
certain nombre de traits agronomiques indésirables. Pour combiner certains traits d’importance
économique du riz asiatique (O. sativa) et du riz africain, des programmes de sélection
interspécifiques ont été lancés par AfricaRice aux débuts des années 1990, ce qui a permis de mettre
au point et d’homologuer plusieurs variétés interspécifiques pour différentes écologies en Afrique.
L’hybridation interspécifique réussie de deux espèces cultivées de riz homologuées et protégées au
travers de la marque déposée New Rice for Africa (NERICA) montre clairement l’utilité du
germoplasme du riz africain dans la mise au point de variétés améliorées modernes qui combinent le
rendement potentiel élevé des parents O. sativa et l’adaptabilité aux différents stress abiotiques et
biotiques des parents O. glaberrima. L’analyse génétique de la collection AfricaRice a permis de
mettre au point un ensemble de base de 1 330 accessions et un mini-ensemble de base de
300 accessions. L’ensemble de base comporte 61 % de l’ensemble de la collection tandis que le mini-
ensemble de base comporte 14 % de ladite collection. Les deux représentent entre 97 et 99 % du
polymorphisme du nucléotide simple (SNP) et pratiquement tous les allèles et les fréquences de
génotype observés dans toute la collection O. glaberrima.
Dans le droit fil des projets visant la préservation des races primitives riches du point de vue génétique
et des variétés traditionnelles au travers de leur culture et de leur commercialisation par les
agriculteurs, certains centres du CGIAR, en collaboration avec des organisations locales et nationales ont
appliqué les outils génomiques pour procéder à une caractérisation génétique de la diversité des races
primitives trouvées dans des collections in situ. Le projet de riz patrimonial des Philippines présenté
dans l’encadré 6 illustre ce point de manière parfaite. Pour les animaux domestiques, un nombre
d´études telles que celle présentée dans l’encadré 7 concernant le chameau de Bactriane a eu recours
aux données de séquence génétique pour appréhender la structure génétique des différentes
populations de races ou de croisements et la relation entre les différentes populations, fournissant de
cette manière des informations utiles pour pouvoir concevoir des stratégies de préservation et
déterminer les priorités quant aux actions à mener pour la préservation, sur la base de l’érosion
génétique dans certaines populations, races ou espèces.
12Encadré 6 : Application de la caractérisation génétique à la conservation in situ et utilisation du riz
patrimonial aux Philippines
Le projet de riz patrimonial qui a démarré en 2014 est soutenu par le Ministère de l’Agriculture des
Philippines et par l’IRRI. Ce projet a pour objet d’améliorer la productivité, d’enrichir l’héritage des
variétés patrimoniales et de donner aux communautés les moyens de cultiver des variétés
patrimoniales dans les écosystèmes marginaux basés sur la culture du riz aux Philippines. Certaines
variétés de riz patrimonial présentent des qualités exceptionnelles en termes de cuisson, de goût,
d’arôme, de texture, de couleur et de valeurs nutritives, d’autres résistent mieux aux maladies et
présentent une grande tolérance aux stress écologiques. Cependant, les agriculteurs doivent
surmonter plusieurs obstacles s’ils veulent cultiver et vendre ces variétés. Parmi ces obstacles, citons
le manque d’accès à des semences de qualité, les faibles rendements, etc. Certaines variétés
patrimoniales disparaissent progressivement. En plus du maintien de la biodiversité dans la région, le
développement des marchés et des produits est crucial pour que les agriculteurs puissent continuer
de cultiver ces variétés de riz menacées. En caractérisant les races primitives existantes, les
scientifiques ont eu recours à l’analyse ADN pour saisir les relations entre les différentes variétés et
pour déterminer la présence de gènes novateurs associés à des traits importants tels que la résistance
aux parasites et aux maladies, ces traits étant susceptibles de protéger ces races primitives uniques
contre ces stress. Ce travail permettra d’augmenter les connaissances des agriculteurs concernant
leur diversité de riz patrimonial et leurs débouchés commerciaux. Dans ce projet, les agriculteurs et
autres parties prenantes ont reconnu l’importance de préserver ces variétés uniques en leur genre
pour les générations à venir. La conservation ex situ est donc poursuivie en vertu d’un accord de
« boîte noire » à l’échelle nationale (Génothèque de l’Institut de recherche rizicole aux Philippines) et
des banques de gènes internationales (Institut international de recherche sur le riz (IRRI).
13Analyse factorielle multiple (AFM) de variétés de riz traditionnel et de riz patrimonial
Analyse factorielle multiple (AFM) effectuée par FactoMineR (paquet R) indiquant les relations entre 119
variétés de riz cultivées in situ par les agriculteurs, sur la base des groupes de variables suivants : (1)
Région des Philippines = Province SOURCE [catégoriel : Région administrative de la Cordillère (CAR,
hautes terrasses irriguées) = Benguet (B), Ifugao (I), Kalinga (K), Mountain Province (MP) et Mindanao
(Région 12, bas-plateaux non endigués) = North Cotabato (NC)]; (2) Génotype, 558 SNP (catégoriel :
A,T,C,G) ; (3) Traits morphoagronomiques, 8 catégoriels + 4 numériques ; (4) Qualité du grain, 1
catégoriel + 21 numériques ; (5) Réaction aux maladies (BB) [catégoriel : résistant (R), moyennement
résistant (MR), moyennement vulnérable (MS), vulnérable (S)].
14Encadré 7 : Appréhension de la diversité génétique du chameau de Bactriane à des fins de
préservation
Comme ils résistent au froid, à la sécheresse et aux hautes altitudes, les chameaux de Bactriane
(Camelus bactrianus) sont particulièrement appréciés dans les steppes et les montagnes de l’Asie
centrale. Ils pourraient être une source de traits potentiellement utiles dans les programmes d’élevage,
mais leur nombre est en décroissance ces dernières années. Les stratégies de préservation et d’élevage
sont entravées, entre autres, par le fait que les connaissances de la diversité génétique des chameaux de
Bactriane et la relation avec des populations existantes sont extrêmement limitées. Avec des partenaires
de recherche situés en Chine et en Mongolie, les scientifiques de l’ILRI ont appliqué l’analyse génétique
en utilisant des marqueurs microsatellites pour caractériser des populations de chameaux de Bactriane
dans ces deux pays. L’étude a révélé des différences notables entre les populations chinoises et les
populations mongoliennes. Elles présentent des flux génétiques au sein des populations cibles (résultant
probablement du commerce effectué le long de la route de la soie, mais aussi de la transhumance).
L’étude a confirmé que les chameaux de Bactriane de Chine et de Mongolie sont éloignés du point de
vue génétique et qu’ils doivent être considérés comme des populations bien distinctes dans les cadres
de préservation et d’élevage (Jianlin et al, 2004).
3. Contribution des informations sur les séquences génétiques dans
l’utilisation de la diversité biologique
Les obligations des parties contractantes quant à l’utilisation durable de la diversité biologique sont
définies dans l’article 10 (utilisation durable) et l’article 7 (identification et surveillance liées à la
conservation et à l’utilisation durable).
Les variations génétiques naturelles ont permis à l’être humain de sélectionner des plantes, des animaux
et des microorganismes durant ces 13 000 dernières années. Grâce à l’application de différentes
techniques et méthodologies d’élevage, l’être humain a été en mesure de modifier le génotype de ces
ressources et de sélectionner des expressions différentielles de traits (phénotype) afin d’obtenir des races
animales, des variétés végétales ou des cultivars et souches de microorganismes capables de répondre à
nos besoins changeants et émergents.
Au sein du secteur agricole, l’information génomique contribue à l’exploitation durable de la diversité
biologique dans le contexte de la sélection végétale et de l’élevage. Dans un sens conventionnel, la
sélection et l’élevage dépendent de la variation génétique naturelle ou induite combinée avec la sélection
efficace de combinaisons génétiques favorables et l’évaluation de phénotypes pour identifier des
variantes d’intérêt pour des traits désirables. La sélection conventionnelle peut être améliorée et elle a
été améliorée, raccourcie et rendue plus précise grâce à l’usage de l’information génomique.
Les économistes estiment que le taux actuel de gain génétique dans l’amélioration des variétés et des
races animales doit être doublé pour répondre à la croissance de la population et des revenus. Les défis
scientifiques à relever sont importants pour pouvoir pleinement mettre au point les technologies qui nous
15Vous pouvez aussi lire

















































