INTRODUCTION À LA MICROSCOPIE DE FLUORESCENCE - FRANÇOIS MICHEL PHD - INMED
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Rev : 10/01/2019
Introduction à la microscopie
de fluorescence
François MICHEL PhD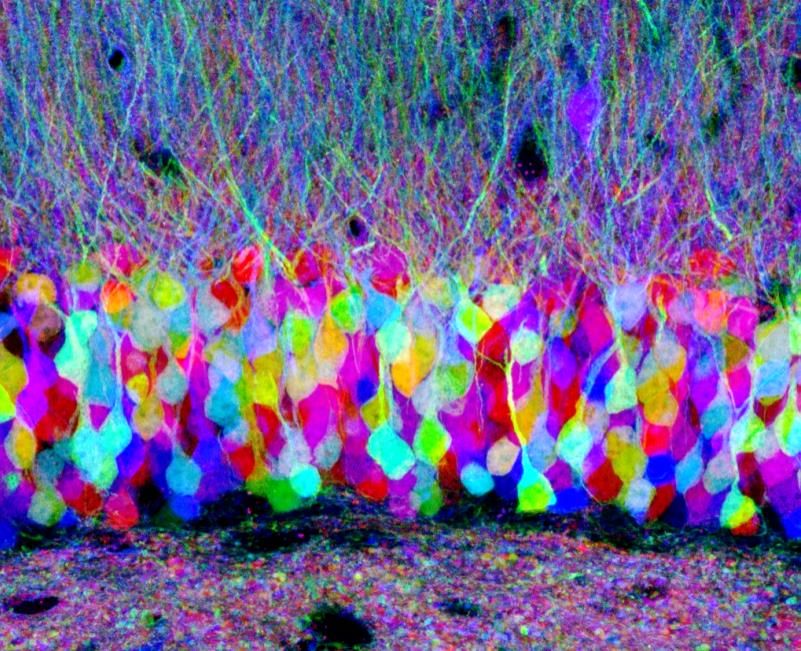
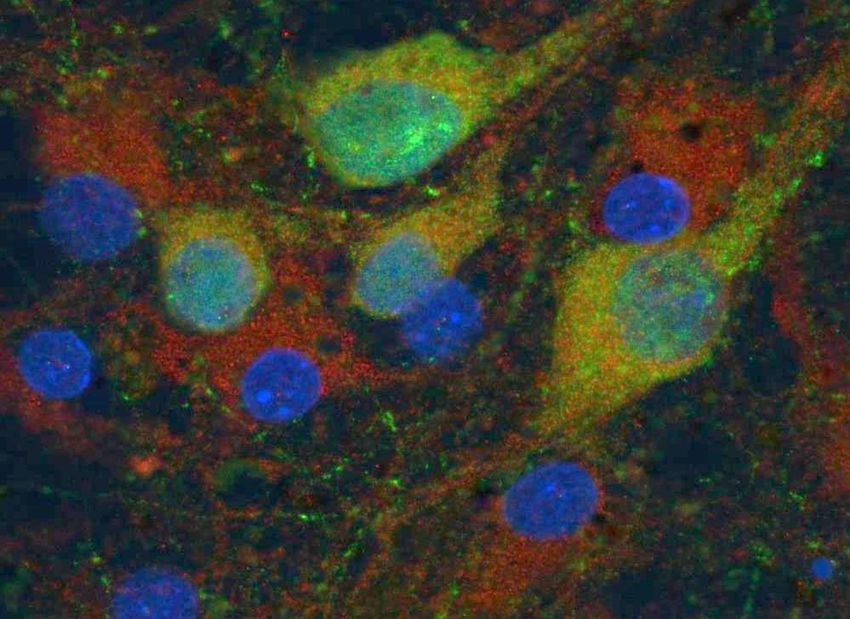
F. Michel 10.01.19 Quelques images en fluorescence
F. Michel 10.01.19
Ce qui vous attend
I. Microscopie en transmission
II. Microscopie de fluorescence
➢ 2.1 Fluorescence ??
➢ 2.2 Microscopie à épi-fluorescence
➢ 2.3 Entrons dans la 3ème dimension
➢ 2.4 Imagerie multiphoton
➢ 2.5 Transparisation et feuillet de lumière
III. Imagerie avancée
F. Michel 10.01.19
Principes de la fluorescence
Perte d’énergie (potentielle,
thermique, cinétique…) : relaxation
État excité (S1) : orbitale électronique extérieure
Emission de
fluorescence
= perte d’énergie
e-
2-Relaxation
S1 (10-12 s.)
1-Excitation
(10-15 s.) 3-Fluorescence
État stable (S0)
Excitation (10-9 à 10-7 s.)
= apport d’énergie
S0
Diagramme énergétique
Molécule fluorescente = Fluorophore (très simplifié) de Jablonski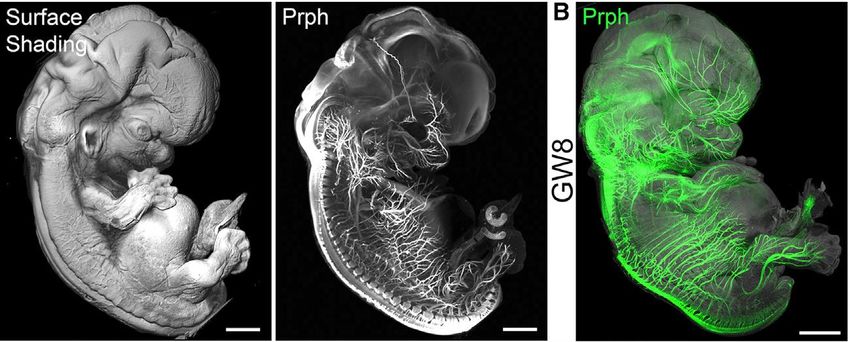
F. Michel 10.01.19
Principes de la fluorescence
L’excitation et l’émission sont des phénomènes quantiques : la molécule
accepte une certaine quantité d’énergie, ni trop ni trop peu, pour être excitée
et réémet cette énergie autour d’une valeur privilégiée (pic d’émission).
L’énergie d’un photon est : Eγ = hc/λ Où h et c sont des constantes
(Planck ; vitesse de la lumière dans
Eγ le vide)
UV 400 500 600 700 IR
S1 Longueur d’onde l (en nm)
488 nm
Les différents états
E vibrationnels et
rotationnels des niveaux
d’énergie se reflètent dans
les spectres d’émission et
S0 d’excitation.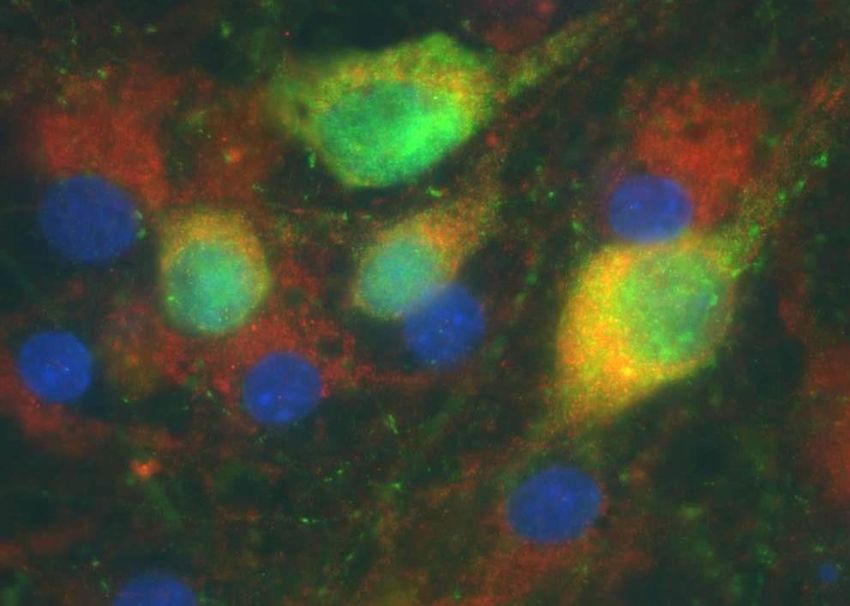
F. Michel 10.01.19
Principes de la fluorescence
Chaque molécule fluorescente a des spectres d’excitation et d’émission qui
lui sont propres (influencés par la structure chimique de la molécule et les
conditions physique (pH, T…)).
Perte d’énergie entre excitation et émission
Décalage vers le rouge : (Stokes shift)
Excitation E1>E2 Emission
Intensité de fluorescence
Pic d’émission
E1 E2
l (nm)
300 400 500 600
Pic d’excitation = efficacité
Plus assez d’énergie
d’acceptation des photons
maximale
pour exciter Exemple : DAPI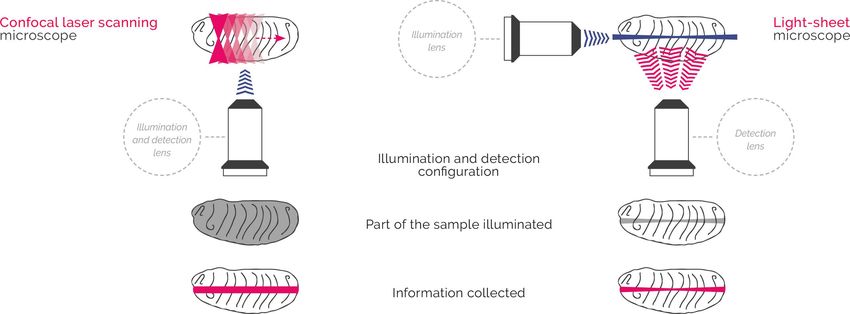
F. Michel 10.01.19
Principes de la fluorescence
L’intérêt majeur de la fluorescence réside
dans son rapport signal sur bruit très
favorable.
On peut ajouter d’autres avantages:
- spécificité du marquage
- compatible in vivo et non toxique
- simple d’utilisation
- peu chère
- versatile…
F. Michel 10.01.19
Protéines fluorescentes
Gènes de méduses ou de coraux optimisées pour
différentes fonctions et caractéristiques telle que :
brillance, spectrale, stabilité, vitesse d’expression, suivi de
l’activité enzymatique, sensibilité au pH, au Ca2+, au Cl- …
Excitation Emission
Max. (nm) Max (nm)
CFP 434 477
GFP 489 508
YFP 514 527
Ds Red 558 583
R Tsien : Nobel chimie 2008 « Brainbow mouse » Livet et al. Nature 2007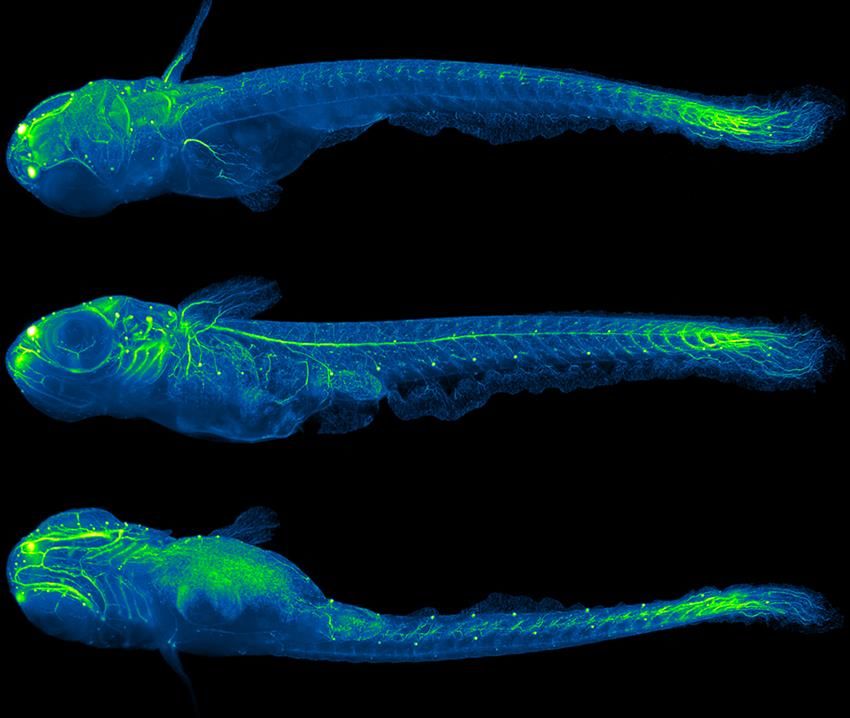
F. Michel 10.01.19
Ce qui vous attend
I. Microscopie en transmission
II. Microscopie de fluorescence
➢ 2.1 Fluorescence ??
➢ 2.2 Microscopie à épi-fluorescence
➢ 2.3 Entrons dans la 3ème dimension
➢ 2.4 Imagerie multiphoton
➢ 2.5 Transparisation et feuillet de lumière
III. Imagerie avancée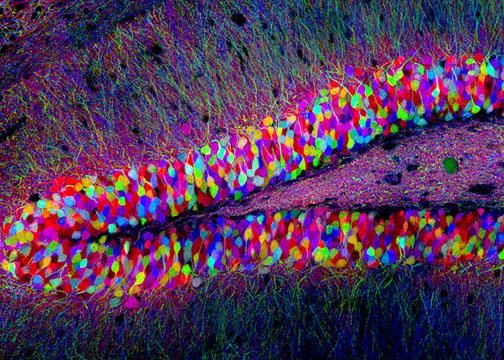
F. Michel 10.01.19
Microscope à épi-fluorescence
Détection
Caméra
Lumière d’émission
Miroir dichroïque
Filtre d’excitation Objectif
Illumination Echantillon
Généralement blanche
(Lampe à vapeur de mercure)F. Michel 10.01.19
Différents types de filtres
transmission xxx nm
xxx nm xxx nm
yy nm
passe-bas l passe-bande l passe-haut l
(LP xxx) (BP xxx/yy) (HP xxx)
BP 525/50 signifie : le filtre
transmet de 500 à 550 nm
X UV IR radio
400 500 600 700 Longueur d’onde l (en nm)F. Michel 10.01.19
Différents types de filtres
DAPI Alexa 488 Alexa 555
ExcitationF. Michel 10.01.19
Différents types de filtres
BP BP
455-495 520-570
Excitation
Passe-bas
LP 365
Cross-talk
BP
Emission BP
505-555
420-460
(440/40) Passe-haut
HP 575
Fluo verte exclue pour éviter
la pollution issue de
l’excitation de l’A555F. Michel 10.01.19
Microscope à épi-fluorescence
Cube à fluorescence
Caméra
Filtre
d’émission
Miroir
dichroïque
Chaque trio (cube) : miroir dichroïque
+ filtres d’excitation et d’émission est
propre aux fluorophores observés
individuellement et en combinaisons
(marquages multiples)F. Michel 10.01.19
Ce qui vous attend
I. Microscopie en transmission
II. Microscopie de fluorescence
➢ 2.1 Fluorescence ??
➢ 2.2 Microscopie à épi-fluorescence
➢ 2.3 Entrons dans la 3ème dimension
➢ 2.4 Imagerie multiphoton
➢ 2.5 Transparisation et feuillet de lumière
III. Imagerie avancéeF. Michel 10.01.19
Bienvenue dans la 3ème dimension
La fluorescence générée hors focus induit
un effet de flou qui altère l’image.
On collecte tous les photons issus de la
colonne d’illumination (diabolo) .
ZF. Michel 10.01.19
Bienvenue dans la 3ème dimension
La fluorescence générée hors focus induit
un effet de flou qui altère l’image.
On collecte tous les photons issus de la
colonne d’illumination (diabolo) .F. Michel 10.01.19
Microscope à Illumination structurée
Caméra
+ +
"Grille"
Réseau de lignes
plan focal de l’objectifF. Michel 10.01.19
Microscope à Illumination structurée
MIS Plein champsF. Michel 10.01.19
Microscope à Illumination structurée
MIS
Plein champsF. Michel 10.01.19
Microscope à Illumination structurée
Z : 43 µm
20 µm
40 X (ON=1.3) cube TR tps d’expo : 15 ms (87 plans DZ=0.5 µm)F. Michel 10.01.19
Microscope
Microscope à Illumination structurée
à Illumination structurée
La MIS peut doubler la résolution : Super-résolutionF. Michel 10.01.19 Microscope à Illumination structurée Avantages de la MIS - Facilité d’utilisation - Sectionnement optique et génération de piles d’images. - Acquisition "rapide"
F. Michel 10.01.19 Microscopie confocale
F. Michel 10.01.19
Microscopie confocale
Illumination
laser Miroir
dichroïque
• L’illumination par lasers
Miroirs de • Illumine l’échantillon par balayage.
• La fluorescence est captée par
scan X-Y
l’objectif puis filtrée (dichroïque et
X Objectif filtres d’émission) et envoyée vers
un détecteur.
Y • La vitesse de balayage, l’énergie et le
pointé du laser jouent sur la qualité
Plan focal de l’image.F. Michel 10.01.19
Microscopie confocale
Détection
PMT
Diaphragme
(Pinhole)
Fluorophore hors focusF. Michel 10.01.19
Microscopie confocale
Les PMT mesurent la quantité de photons
(quelque soit l) en fonction du temps Détection
(échantillonnage temporel) et la taille du pixel PMT
correspond donc au temps t d’intégration du
signal.
Au temps t1 le point de scan de coordonnée
(x1y1) la fluo a une intensité F1, au temps t2
(x2y2) la fluo est F2... L’image est représentée
pixel par pixel avec les valeurs Fn (XnYn).
Un PMT est un détecteur qui transforme
les photons en électrons et qui amplifie le
courant généré par une forte différence de
potentiel entre la photocathode d’entrée
et l’anode de sortie (+ de 1000 V).
L’amplification est due à l’utilisation
d’électrodes intermédiaires (dynodes)
portées à des potentiels positifs croissants
qui émettent une gerbe d’électrons à
l’arrivé de chaque photo-électron incident.F. Michel 10.01.19
Microscopie Confocale (résolution)
Microscope à fluorescence Microscope confocal
Resxy=0.61l/ON Resxy=0.4l/ON
Resz=2l/ON2 Resz=1.4l/ON2
Résolution max en xy 220 nm Résolution max en xy 130 nm
Résolution max en z 600 nm Résolution max en z 350 nm
Plus l’ON est grande, meilleure est la résolutionF. Michel 10.01.19
Microscopie Confocale
(échantillonnage axial)
1 1
2 2
3 3
1 4
2
Il ne faut pas confondre résolution et
3 épaisseur de coupe optique
Z=f(ON, n, l, pinehole…)
4
Le pas optimal de déplacement en Z (DZ)
2
5 est fonction de la résolution (Resz=1.4l/ON )
et des lois de l’échantillonnage :
6 1/2 Resz< DZF. Michel 10.01.19
Pixels et Voxels …
“Picture element” et “volumetric element” ; échantillonage
Z
Y X
Z>> X=Y X>> Y=Z
Y=X et Z=0F. Michel 10.01.19
Microscopie confocale multidimensionnelle
XY Scan XY Scan
X
Y Z
X
Temps XY et Z Scan
Y
Les dimensions peuvent être : la profondeur Z, le temps T,
la couleur l … On peut donc faire des images 3, 4 voir 5DF. Michel 10.01.19
Limitations de la microscopie confocale
➢ Pénétration limitée dans la profondeur du tissu épais
(F. Michel 10.01.19
Microscopie Confocale à spinning disk (Nipkow)
➢ Pénétration limitée dans la profondeur du tissu épais (50 Hz caméras EMCCD)F. Michel 10.01.19
Ce qui vous attend
I. Microscopie en transmission
II. Microscopie de fluorescence
➢ 2.1 Fluorescence ??
➢ 2.2 Microscopie à épi-fluorescence
➢ 2.3 Entrons dans la 3ème dimension
➢ 2.4 Imagerie multiphoton
➢ 2.5 Transparisation et feuillet de lumière
III. Imagerie avancéeF. Michel 10.01.19
Principes physiques de la
microscopie multiphoton
Relaxation Relaxation
S1 État virtuel g
S1
Excitation
Excitation
g g
Fluorescence Fluorescence
S0 S0
Source Laser
(continue)
Eg = hc/l LASER IR pulsé
(femtoseconde)
État virtuel signifie une fenêtre temporelle de 10-18 secondes
(une attoseconde) et spatiale de l’ordre de 10-12 cm3 (nuage
électronique).
(théorie par Maria Goppert-Mayer
1929 (prix nobel 1963))F. Michel 10.01.19
Principes physiques de la
microscopie multiphoton
les conditions nécessaire à l’excitation bi-photonique impliquent une très haute densité
de photons que ce soit d’un point de vue spatial et temporel, conditions remplies par
certain lasers pulsés.
Faisceau continu
Puissance moyenne de 1 à 200 mW
12 500 ps
Faisceau pulsé
Puissance moyenne de 1 à 5 W
= 0,1 ps
P : puissance moyenne
Probabilité d’absorption bi-photonique : A2g = P2/ (.F) : durée du pulse
F : fréquence
.w>K donc pour 100 fs à 800 nm on a un pulse de 6,7 nm
Pour 20 fs le pulse est de 33,6 nmF. Michel 10.01.19
L'excitation bi-photonique est
restreinte au point focal
1 photon 2 photons
1 photon 2 photons
Le balayage induit une coupe
optique intrinsèquement confocale,
Point focal inutile d’enlever les photons hors
focus (non générés).F. Michel 10.01.19
Longueurs d’onde d’absorption optimales
Le spectre d’excitation biphoton
n’est pas forcement le double
point à point du spectre
d’excitation monophoton
(optique non-linéaire).
Excitation 3- photons
Exemple : DAPI
Utilisation d’un laser
Dont la longueur
d’onde est fixe à
1030 nm : T-pulse
(Amplitude
Systems).
F. MICHEL 2005F. Michel 10.01.19
Imagerie bi-photon vs confocale
Faibles profondeurs : objectif 63X à huile
Confocal
-meilleure
résolution
-meilleur rapport
signal/bruit
20 µm 40 µm 60 µm 80 µm
Bi-photon
-plus de bruit
-coupes optiques
plus épaisses
F. MICHEL 2005F. Michel 10.01.19
Imagerie bi-photon vs confocale
Pour des profondeurs plus importantes : objectif 63X à eau
Confocal
- augmentation de la puissance
du laser
- augmentation du bruit
100 µm 120 µm
Bi-photon
- augmentation
de la puissance
du laser
- acquisition plus
facile qu’en
confocal F. MICHEL 2005
100 µm 140 µm 170 µm 200 µm
Fin de la tranche…F. Michel 10.01.19
Limites du multiphoton
• Les fenêtres optique 700-900nm et 1100-1300nm sont optiquement
transparente
• Echauffement dans le domaine d’absorption de l’eau =>formation de
bulles, éclatement de l’échantillon.
F. MICHEL 2005
Spectre d’absorption de l’eau
Immuno and Cell Bio 2010, 88(4):438-44F. Michel 10.01.19
Avantages du multiphoton
AVANTAGES
- Volume d’excitation plus faible (intrinsèquement confocale)
- Élimination quasi-totale de la fluorescence hors plan focal
- Moindre photo-blanchiment général et photo-toxicité
- Pénétration plus grande des IR dans les tissus (jusqu’à 1 mm)
- Excitation des fluorophores UV sans lumière UV
- Bonne séparation des lumières d’excitation et d’émission
INCONVENIENTS
- Nécessité d’un important flux de photons
- Balance entre puissance et effets thermiques
délicate
- Coûts et maintenance de l’appareillage lourd
- Pour plus de détails voir film chapitre 3.2F. Michel 10.01.19
Ce qui vous attend
I. Microscopie en transmission
II. Microscopie de fluorescence
➢ 2.1 Fluorescence ??
➢ 2.2 Microscopie à épi-fluorescence
➢ 2.3 Entrons dans la 3ème dimension
➢ 2.4 Imagerie multiphoton
➢ 2.5 Transparisation et feuillet de lumière
III. Imagerie avancéeF. Michel 10.01.19
Pourquoi "transpariser" les tissus?
• La Relation structure/fonction en 3D d’échantillons
biologiques est riche d’information
• Nous devons améliorer l’exploration des tissus et des
organes au plus proche de la morphologie in vivo à
des résolutions mésoscopique (du µm au cm)
• plusieurs phénomènes empêchent la transmission
correct de la lumière dans le tissu (diffusion…)F. Michel 10.01.19
Pourquoi "transpariser" les tissus?
Wilhlem Conrad Röntgen
La première radio par rayons X
Prix Nobel de physique en 1901 pour sa
découverte des rayons X qui peuvent
traverser le corps humainF. Michel 10.01.19
Un peu plus de physique
Comme les rayons X, la lumière est une onde électromagnétique qui
interagie avec la matière par :
- Absorption (mélanine, hémoglobine…)
- Diffusion / diffraction qui induisent un retard de propagation de
l’onde lumineuse représenté par l’indice optique n ou RI.
Soit c = la vitesse de la lumière dans le vide
On a n = la vitesse de propagation de l’onde dans le milieu conducteur
n = c/n et n≥1 Milieu n @ 560 nm
eau 1.33
n est proportionnel à la densité de Proteines 1.6
molécules et d’électrons qui vont interagir Lipides 1.46
avec l’onde. Les lipides ont une forte densité Mélanine 1.7
électronique et ralentissent plus la lumière Cytoplasme 1.37
que l’eau (nlipides = 1.46)
Membrane 1.48
Noyau 1.39F. Michel 10/01/2019
Un peu plus de physique
En pratique, un milieu homogène ne diffuse pas de lumière
L’objectif est d’homogénéiser les indices de réfraction dans les
tissus et d’éliminer les points de diffusion de la lumière. Le RI du
milieu peut être augmenté et/ou le RI des tissus diminué.F. Michel 10.01.19
Comment "transpariser" les tissus?
Werner Spalteholz
Anatomiste
(1861-1940)
Belle M. et al 2017 CellF. Michel 10.01.19
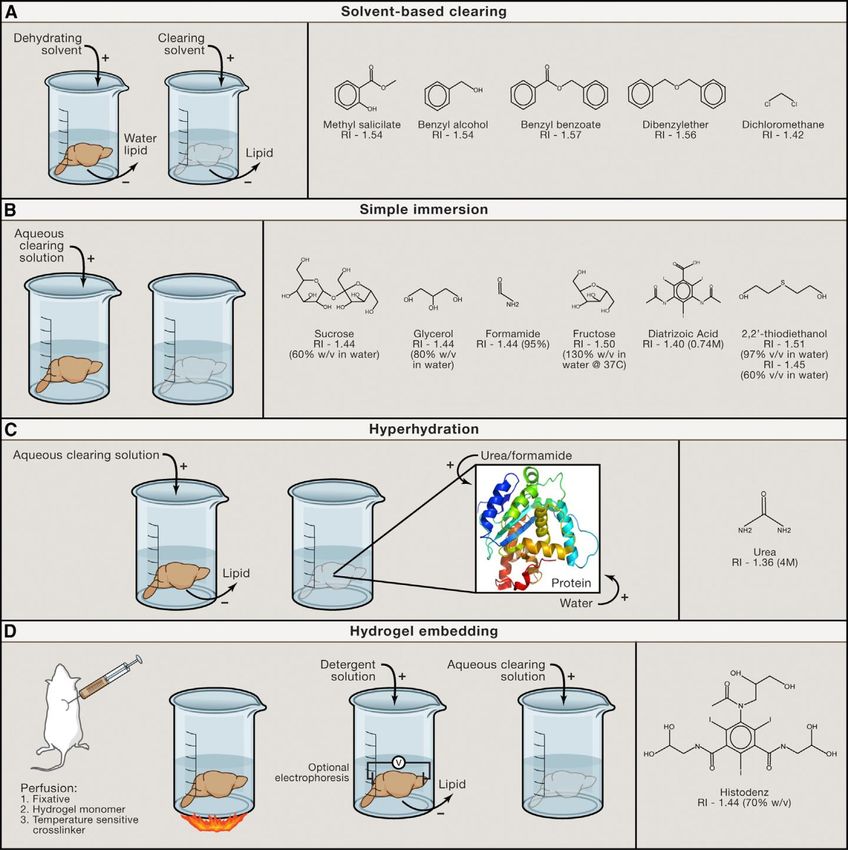
Comment "transpariser" les tissus?
Solvents
(3/i/U)-DISCO - BABB
Simple immersion
Sucrose, Focus
clear, SeeDB
Hyper-hydratation
Urée, Scale, CUBIC
Intégration dans un
hydrogel
Clarity, PACTF. Michel 10.01.19
Clarification
Procédure de clarification :
Clarity, I-DISCO, CUBIC,…
Milieu inhomogène,
Milieu homogène, tissus clarifiés
tissus biologiques classiques
Amélioration de la pénétration et
réduction de la diffusion de la lumière,
conservation de la fluorescence, …F. Michel 10.01.19
Observations (épi-fluorescence)
Tdtomato
CLARITY Oxytocin-GFP,
CUBIC Oxytocin-GFP, CUBIC
Tranche de tronc
cérebral (1mm)
multi-photon Confocal
3 mm (Trimscope II)
Baude A, Tressard T, Matarazzo V, Felix MS … INMEDF. Michel 10.01.19
SPIM : la lumière en feuillet
Avantages du SPIM :
• Résolution axiale (1 à 10 µm)
• Fort ratio signal/bruit (pas de signal hors
focus) ; peu de photo-bleaching et de
photo-toxicité
• Très rapide (jusqu’à 200 fps)
Pour revue lire : Huisken J et al 2012 Development • Grand champs et possibilité de tracking
• PrixF. Michel 10.01.19
Observations (feuillet de lumière)
High Photobleaching Low
Slow Imaging speed Fast (1000x)F. Michel 10.01.19
Observations
Microscopie
Microscopie
à feuillet de lumière Choix du microscope dépendant confocale
de la question scientifique
Vitesse d’acquisition, photobleaching , grossissement,
Résolution spatial, détection de photons…F. Michel 10.01.19
Analyses à grande échelle
Compter les cellules ou quantifier les intensités du signal, dans une région
anatomique précise en utilisant les informations d’annotation associées à un atlas
(Baude A, Bollmann Y… INMED)F. Michel 10.01.19
Sources et Ressources
WEB divers
➢ Molecular expressions http://micro.magnet.fsu.edu/index.html
➢Microscopy U (Nikon) : http://www.microscopyu.com
➢Microscopy resource system (Olympus): http://www.olympusmicro.com/index.html
➢A whole world of microscopy knowledge (Zeiss): http://zeiss-campus.magnet.fsu.edu/index.html
➢Wikipédia
Plate-formes d’imagerie
➢ PICSL http://www.picsl.univ-mrs.fr/ : MICROSCOPIE OPTIQUE (PDF indisponible)
➢ BIC http://www.bic.u-bordeaux2.fr/ : Microscopie à épi-fluorescence et microscopie confocale (PDF)
➢Membres du GDR2588 CNRS (microscopie fonctionnelle des systèmes vivants) et
du RTmfm (Réseau technique de microscopie de fluorescence multidimensionnelle).
Cours
➢ Arnaud Sergé (université de la Méditerranée – Marseille II)
➢Yves Husson (Université Joseph Fourrier – Grenoble)
➢Serge Monneret (Institut Fresnel, Marseille)
➢Maxime Dahan (Laboratoire Kastler Brossel, ENS)
➢François Waharte (institut Curie, UMR144)
Handbooks
➢Fondamentaux d’optique et d’imagerie numérique à l’usage des microscopistes (C Cibert, ed. Cépaduès)
➢Handbook of biological confocal microscopy, third edition (JB. Pawley ed. Springer)
➢Fundamentals of light microscopy and electronic imaging (DB. Murphy, ed Wiley-liss)
Commentaires, remarques, améliorations :
francois.michel@inserm.frVous pouvez aussi lire

















































