La Neuro-Myélite Optique en 2013 - Dr DA Laplaud CHU de Nantes Inserm CRC1064
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
La Neuro-Myélite Optique en 2013
Dr DA Laplaud
CHU de Nantes
Inserm CRC1064
Cours de DES 29/03/2013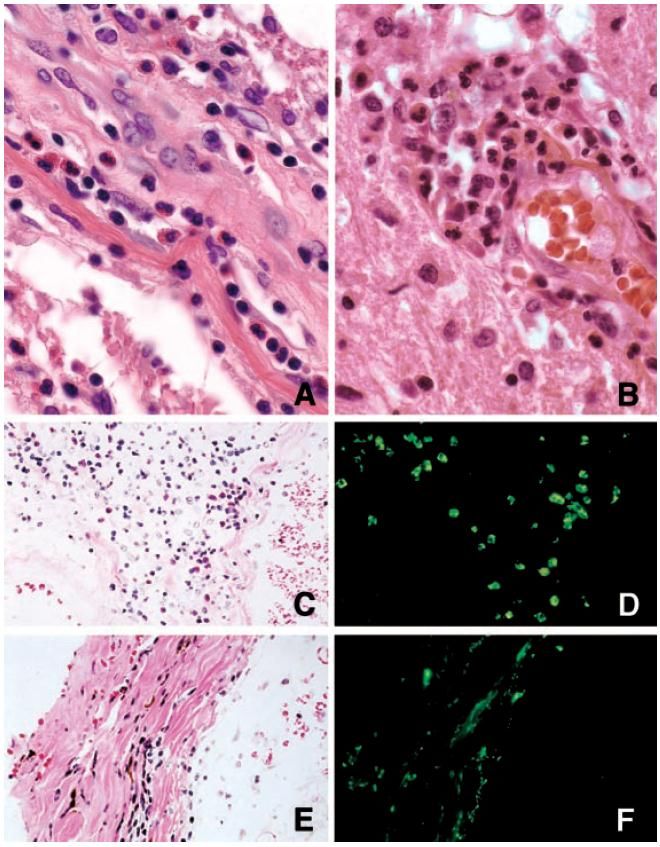
La NMO Définition : Maladie rare inflammatoire et démyélinisante du SNC, responsable d’une atteinte visuelle (NORB) et médullaire (myélite), monophasique ou récurrente. Un peu d’historique : Maladie décrite par Gault et Devic en 1894 sur 16 cas de la littérature et un de leur patient. La maladie était alors décrite comme une succession rapide de névrite optique bilatérale et une myélite. Par la suite, des cas moins sévères ont été inclus. Une forme frontière de SEP a été longtemps discutée, mais la maladie s’en éloigne par plusieurs caractéristiques pathologiques, radiologique et immunologiques .
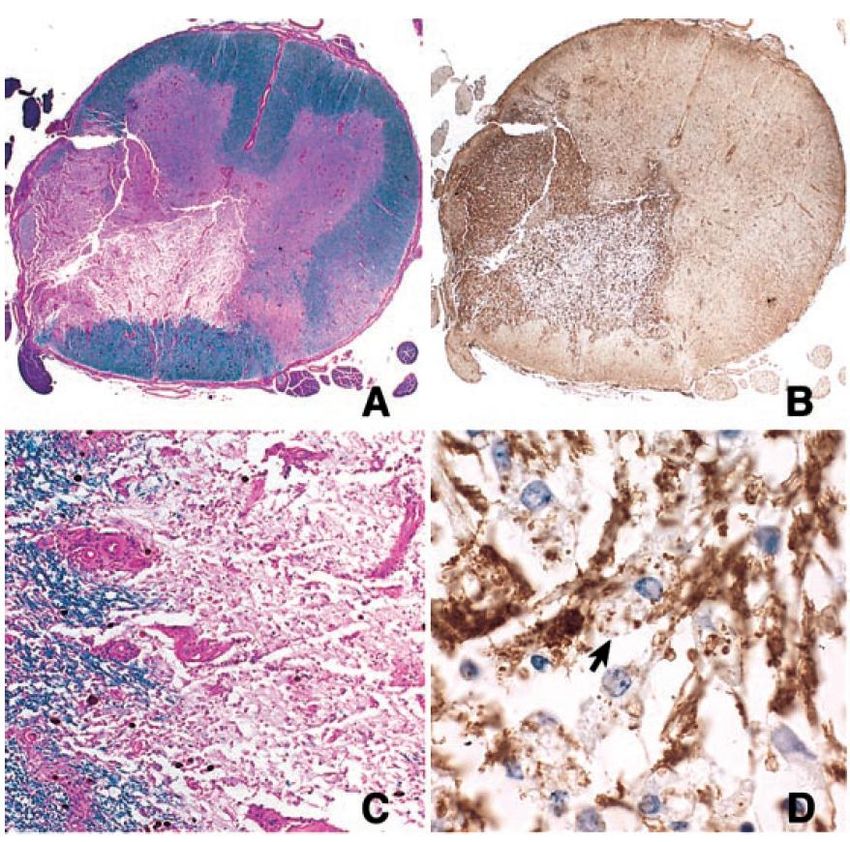
Neuropathologie
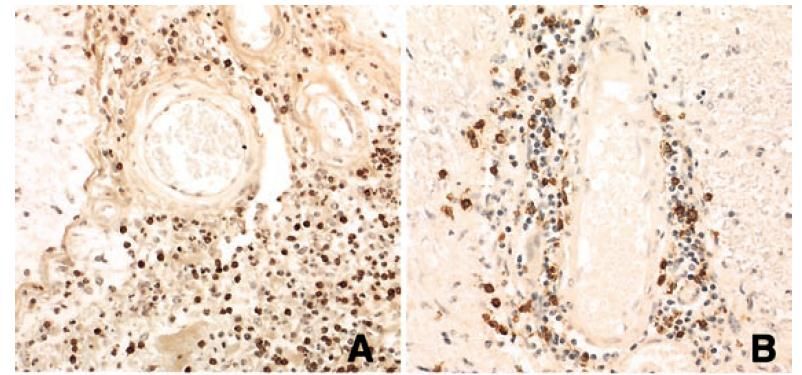
La NMO
20 et 21 juin 2007 – Nantes
•Dépôt de Complément activé.
•NMO : une pathologie dysimmunitaire humorale touchant les
régions périvasculaires.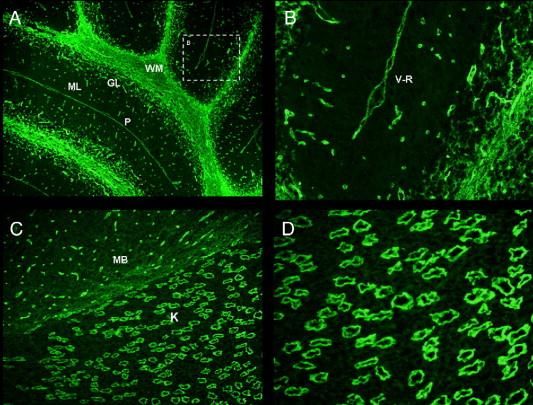
Les anticorps anti-AQP4 •Cible antigénique :AQP4 •AQP : water channel. Sert au transport de l’eau à travers les membranes plasmiques. AQP4 surtout au niveau du SNC. •NMO-IgG se lie au structures vasculaires dans le SNC, partie distale des tubes collecteurs dans le rein et cellules pariétales de l’estomac. •Molécule exprimée sur les terminaisons astrocytaires
Les anticorps anti-AQP4 •Recherche d’auto-anticorps : mise en évidence d’un Ac dans le sérum des patients se fixant sur le cervelet de souris. Spécifique de patients avec NMO : NMO-IgG. •45 patients avec NMO (33 + pour NMO-IgG), 19 avec SEP (2 +) et 56 contrôles (0 +) testés : sensibilité 73%, spécificité de 91 %. •Différentes études publiées : sensibilité env. 50 %. •Amélioration de la sensibilité au cours du temps: env 75% •D’autres patho auto-immunes peuvent avoir des NMO-IgG + lorsque atteinte du NO ou moelle (SLE, Sjogren, SEP asiatiques)
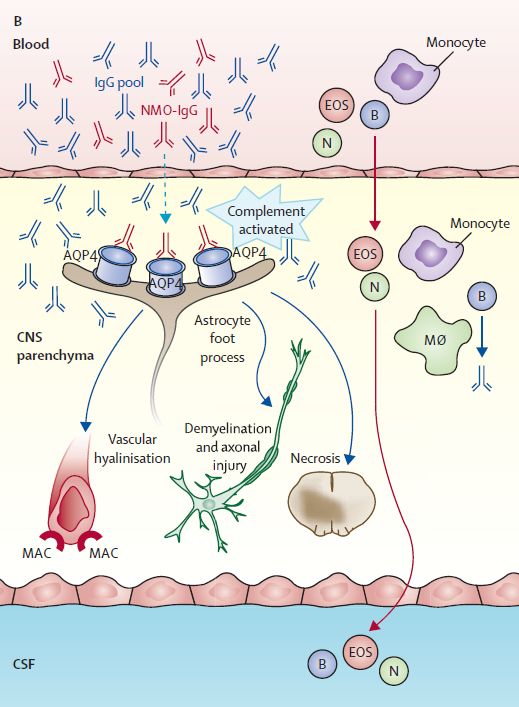
La physiopathologie Weinshenker et al, Lancet Neurol, 2007
Les critères diagnostiques

Le spectre de la NMO
Le spectre de la NMO
Les anti-AQP4 prédisent la survenue d’un 2ème evt
Weinshenker et al, Ann Neurol, 2006
Lennon et al, Neurology, 2008Epidémiologie •Sex Ratio de 3:1 à 9:1, F>>H. Sauf pour les formes monophasiques. •Moyenne d’âge 34-39 ans. Des cas pédiatriques existent. •Atteint plutôt les non-caucasiens (hispaniques, Afro-américains, asiatiques) •Incidence et prévalence difficile à estimer car maladie sous- diagnostiquée. Env. 1-3% des maladies démyélinisantes. •Attention aux formes optico-spinales de SEP qui sont différentes (formes asiatiques). Overlap ou sous-groupe.
Eléments Cliniques •Association dans le même temps ou de façon consécutive d’une NORB et d’une Myélite Aiguë Transverse •NORB plus sévère que dans la SEP, laissant souvent des séquelles. •MAT différente de la SEP car complète et bilatérale avec paraparésie ou paraplégie, niveau sensitif et dysfonction sphinctérienne. Peuvent s’ajouter des douleurs pseudo-radiculaires et des dysesthésies. •Description de cas atypiques: hoquets incontrôlables, limb shaking précedant l’atteinte médullaire. •Après le premier évènement définissant la NMO soit il n’ y a pas de nouveau symptôme (monophasique) soit des poussées de NORB ou MAT peuvent survenir séparées de plusieurs mois ou années (forme récurrente, 80 %). •Autres maladies auto-immunes associées : thyroïdite, SLE, Sjogren. •Fréquemment patients positifs pour SSA ou FAN, en l’absence d’atteinte systémique.
Epidémiologie Collongues et al, Neurology, 2010
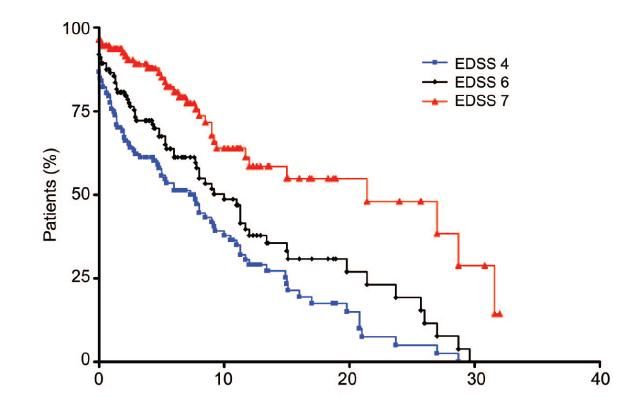
Epidémiologie
Tps moyen pour EDSS 4: 7 ans
Tps moyen pour EDSS 6: 10 ans
Tps moyen pour EDSS 7: 20 ans
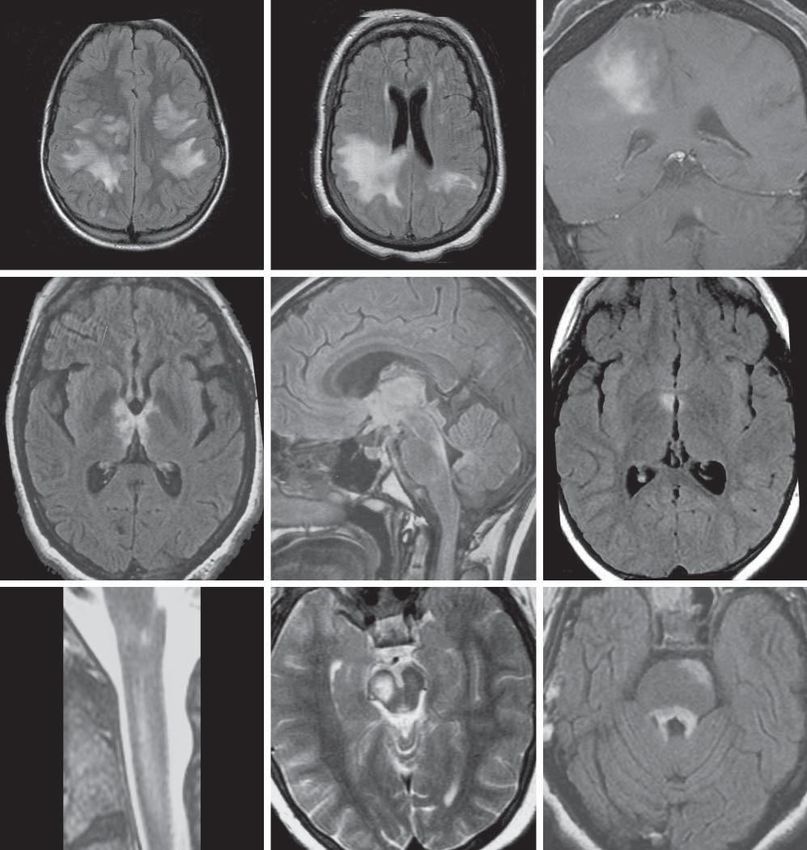
Collongues et al, Neurology, 2010imagerie •Lésion médullaire centrale s’étendant sur au moins 3 segments vertébraux. •Œdème •Lésion Gadolinium positive •Cerveau : lésion du NO au moment de la poussée pouvant s’étendre jusqu’au chiasma. •Habituellement pas d’autres lésions dans le cerveau (ancien critère d’exclusion diagnostique). •Parfois, des lésions du tronc cérébral, du diencéphale ou de l’hypothalamus peuvent être retrouvées. •Des lésions non-spécifiques (n’ayant pas les critères de Barkhoff) peuvent être visualisées.
imagerie 20 et 21 juin 2007 – Nantes
imagerie
imagerie 20 et 21 juin 2007 – Nantes
imagerie
Le LCR •Pendant les poussées : hyperprotéinorachie, Pleïocytose (10%) avec 50-1000 c/mm3. parfois dominé par Neutrophiles. •Parfois présence d’eosinophiles. •BOC90% dans la SEP)
Les traitements •Aucun essai thérapeutique réalisé pour traitement des poussées ou leur prévention •Poussée : 1g/j pdt 5 jours Solu. Plasmaphérèses si inefficace (efficacité démontrée dans un essai en double aveugle sur 19 patients). •TT de fond : à débuter le plus tôt possible •Immunosuppresseurs >> Immunomodulateurs •Azathioprine (3 mg/kg/j) +/- prednisone (1 mg/kg/j) •MMF (1-3 g/j) – 1 essai rétrospectif sur 24 patients •Mitoxantrone (1 essai sur 5 patients), Endoxan •Rituximab (1 essai sur 25 patients) •Attention: possible aggravation sous IFN et natalizumab
Cas clinique
Mme FG, 44 ans
ATCD: depuis un an (1992), présente par « crises » des douleurs à type de
brûlure dans l’hémicorps Dt accompagnées de phénomènes toniques dans
l’hémicorps G. Traité et amélioré par Dépakine.
Notion de piqure de tiques possibles.
1993: Apparition sur 8 jours de douleurs radiculaires bilatérales niveau T6 puis 2
jours plus tard, de troubles sensitifs progressivement ascendant et troubles de la
marche.
Adressées aux Urgences:
Ex Général normal. Constantes normales.
Paraplégie quasi-complète
Troubles sensitifs profonds des M. Sup et Inf.
TVS
Dans le service:
Paraplégie flasque avec BBK bilatéral
Niveau sensitif C3IRM : Odème médullaire étendu sur l’ensemble du cordon médullaire cervical Hypersignal T2 centro-médullaire de la région bulbaire à la région dorsale. Gado- IRM encéphalique normale PL: 120 elt, 60% PNN, pas de BOC Sérologies virales négatives, Lyme négatif
TTT par CTC : amélioration en 48h. IRM de contrôle à 1 mois: Diminution de l’œdème médullaire. Images identiques en T2. IRM à 3 et 6 mois: Hypersignal T2 de C4 à C7. Hyposignal T1 même territoire. Gado-.
Diagnostic de méningo-myélite d’origine indéterminée.
Récupération quasi-complète en quelques semaines/mois.
1995- 2 ans plus tard: NORB OG sévère (amaurose).
5 bolus de corticoïdes.
PL: normale. Pas de BOC.2001 puis 2002: deux poussées motrices sur les membres inférieurs. Apparitions de douleurs suspendues. Garde des séquelles motrices sur le M. Inf D. PM limité mais sup 1 km. 2003: EDSS à 3.5 IRM : Lésion en HST2 linéaire pos de C2 à C6, HST2 en C7, , T2 et T9. Gado- Mise sous IFNb Fin 2004: dégradation du PM qui reste >1km. EDSS stable. IRM: Apparition d’une lésion en C7 Gado+. Solu. Pas d’amélioration à 2 mois. Discussion de Mitoxantrone mais récusée car finalement récupère.
Fin 2005: Poussée médullaire avec paraplégie. Solumédrol avec récupération incomplète. EDSS à 6.5 à 1.5 mois. Puis nouvelle poussée médullaire: déficit moteur complet du M. inf D IRM: nouvel HST2 de T5 à T7, gado+
Mise sous mitox. Stabilisation. Mise ss Cellcept
Vous pouvez aussi lire