Physiopathologie et traitements de la Sclérose Latérale Amyotrophique - Prof. Emmanuel Hermans
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous

Physiopathologie et traitements de la
Sclérose Latérale Amyotrophique
Prof. Emmanuel Hermans
Neuropharmacologie
Institut des Neurosciences
UCL-Bruxelles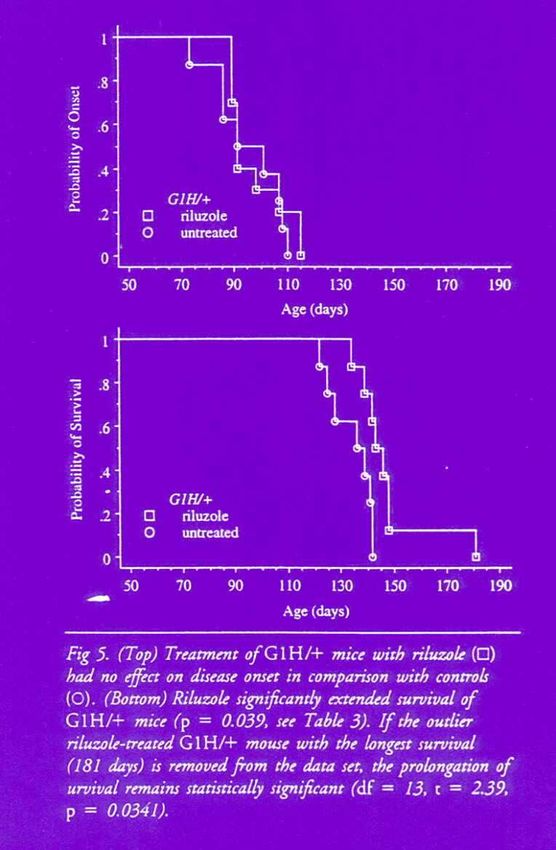
Sclérose latérale amyotrophique
Maladie de Charcot
Syndrome de Charcot
Sclérose de Charcot
Lou Gehrig's disease
Stephen Hawking Lou Gehrig Dr Jean-Martin Charcot
(1942- ) (1903-1941) (1825-1893)
3
Sclérose latérale amyotrophique
Déclin de la qualité de la frappe de Lou Gehrig
durant la saison de baseball 1938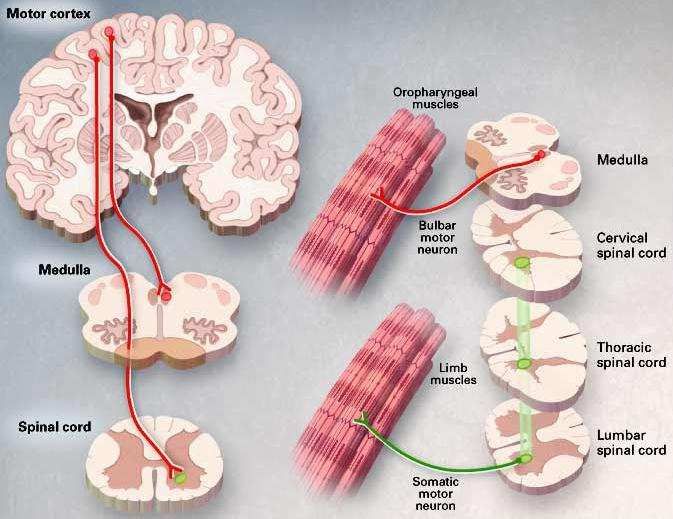
Maladie de Charcot
Amyotrophic Lateral Sclerosis The Lou Gehrig Disease
• Dénervation des muscles
squelettiques amyotrophie
• Perte progressive du contrôle des
activités motrices paralysie
• Systèmes non-moteurs sont
préservés (fonctions intellectuelles
et sensorielles)
• Sphincters et oculomoteurs
préservés!
• Prevalence 2-5/100,000
• Evolution : handicap progressif;
• Maximal incidence 65 years mort par détresse respiratoire
après 3-5 ans
« a mind trapped in a body »
• Pas de traitement efficace à ce jour
La sclérose latérale amyotrophique :
Physiopathologie
Maladie de Charcot
Sclérose latérale amyotrophique The Lou Gehrig Disease
Perte des motoneurones supérieurs (UMN)
spasticité et hyperréflexie Perte des motoneurones spinaux
(LMN) dénervation des muscles
squelettiques, fasciculation et atrophie
Rowland and Schneider, NEJM 322:1688 (2001)
Sclérose latérale amyotrophique
Sclérose latérale amyotrophique
The complex molecular biology of amyotrophic lateral sclerosis (ALS).
Redler RL, Dokholyan NV.
Prog Mol Biol Transl Sci. 2012;107:215-62.
Sclérose latérale amyotrophique
Symptômes essentiellement moteurs :
perte progressive habilité de - marcher - se nourrir- se laver
- parler - déglutir
EVOLUTION
• progression Inexorable
• PAS de récupération
• hétérogène : pas de facteur pronostic vrai
très rapide…. à très lentement évolutive
Stephen Hawking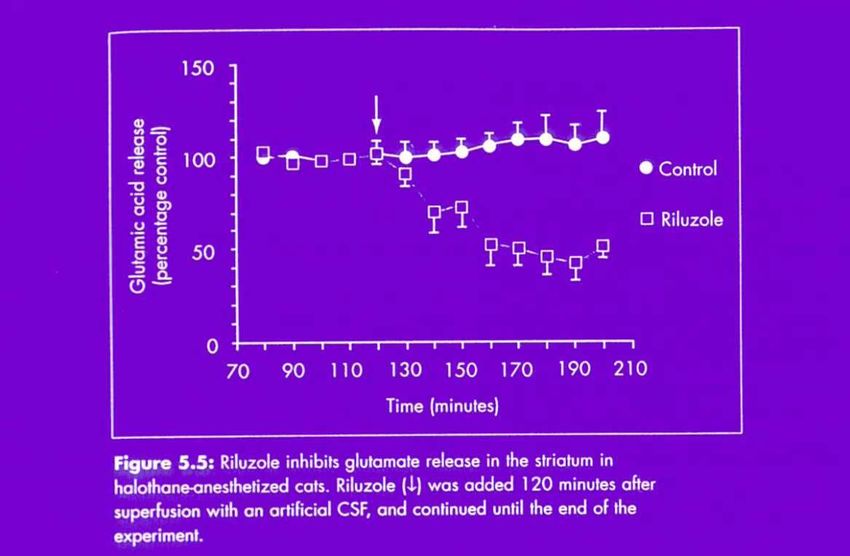
Sclérose latérale amyotrophique
LESIONS
Prédominance des lésions
• région lombaire atteinte membre inférieur
• région cervicale atteinte membre supérieur
• tronc cérébral atteinte bulbaire, muscles cou et thorax
EVOLUTION vers une forme complète !
SIGNES CLINIQUES
• début insidieux vers 60 ans
• délai entre début symptômes & diagnostic
• évolution moyenne = 2 ans, Parfois jusqu’à 10 ans
Sclérose latérale amyotrophique
SIGNES CLINIQUES
1) Atteinte des motoneurones
• 1er symptôme moteur = faiblesse musculaire asymétrique
: - une jambe qui traîne
- difficultés pour écrire
• atrophie musculaire
• fasciculations ! très évocateur
• crampes
s’aggravent progressivement déficit atrophie
Sclérose latérale amyotrophique
SIGNES CLINIQUES
2) Atteinte des voies motrices pyramidales :
• syndrôme pyramidal (perte contrôle motricité volontaire)
3) Atteinte du tronc cérébral (forme bulbaire) : + rare
trouble déglutition
dyspnée
troubles de l’élocution, la phonation, la déglutition, la mimique,
bavage
+ grave car fonction vitales
Sclérose latérale amyotrophique
COMPLICATIONS :
1. Chute fracture convalescence lente & difficile
2. Difficultés progressives déglutition : ? gastrostomie ?
• fausse déglutition & infections respiratoires
• toux
• déshydratation
• perte de poids (favorise aussi l’infection)
3. Difficultés/insuffisance respiratoires : ventilation ?
4. Fatigue
La sclérose latérale amyotrophique :
Prise en charge… en 2014
Sclérose latérale amyotrophique
PRISE EN CHARGE :
1. Rilutek (50 mg 2 x par jour)
• inhibition libération du glutamate
• ralenti l’évolution maladie
• pas de guérisson
• pas d’effets sur les symptômes
• effets 2aires : fatigue et nausées
• hépatite médicamenteuse
Riluzole
PK26124 (Pharmuka, 1980) R54274 (Phone Poulenc, 1983) Rhone
Poulenc Rorer (1991) Aventis (1999) Sanofi-Aventis (2004) Sanofi
(2011) …
Riluzole : propriétés antiépileptiques, anesthésique local, neuroprotecteur
Etude de la neuroprotection : 1989, Rhone Poulenc, choix de la SLA (évolutivité
rapide)
Etudes cliniques 1990-1996 • Riluzole R57274, AG216 1990-92
• Riluzole R57274, RP301-302 1992-94
• Riluzole R57274, AG420 1995-96
• AMM 10 juin 1996 (Belgique)
Riluzole : inhibition de recapture de glutamate,
inhibition de canaux sodiques
L’ efficacité (modeste) du riluzole rejoint la théorie glutamatergique de la
SLA
Le Riluzole en 2014 (Rilutek®)
– ALS : Confirmation d’un effet neuroprotecteur léger
• Cochrane Review 2012
– Pas d’effet symptomatique
– Bien toléré
– Patients répondeurs/non répondeurs
– Freine l’extension des lésions (?)
– Pas utile dans d’autres maladies dégénératives
(plusieurs études encore en cours)Sclérose latérale amyotrophique PRISE EN CHARGE : 1. Rilutek 2. Kiné : • pas rééduquer mais réadapter • mobilisation : maintenir fonctions • prise en charge des symptômes respiratoires • aide expectoration, aide à la toux • confort, installation confortable du patient
Sclérose latérale amyotrophique
PRISE EN CHARGE :
1. Rilutek
2. Kiné
3. Logopédie :
- aide à la déglutition, bonne position
- adapter alimentation éviter textures mixtes
épaissir liquides
- aide à maintenir communication
Sclérose latérale amyotrophique
MAINTENIR LA COMMUNICATION :
1. tableau avec lettre alphabéthique + pointeur laser
2. tableau photos d’action de la vie de tous les jours
3. tableau photos des membres de la famille et proches
4. téléphone + amplificateur de voix
5. communication par ordinateur + synthétiseur de voix
6. clignement des yeux, mouvements des doigtsSclérose latérale amyotrophique
PRISE EN CHARGE :
1. Rilutek
2. Kiné
3. Logopédie :
4. Prise en charge des symptômes
SLEROSE LATERALE AMYOTROPHIQUE (SLA)
Prise en charge des symptômes
Clinical care of patients with amyotrophic lateral sclerosis
The Lancet Neurology, Volume 6, Issue 10, October 2007, Pages 913-925SLEROSE LATERALE AMYOTROPHIQUE (SLA) Clinical care of patients with amyotrophic lateral sclerosis The Lancet Neurology, Volume 6, Issue 10, October 2007, Pages 913-925 SLEROSE LATERALE AMYOTROPHIQUE (SLA) Prise en charge des symptômes Clinical care of patients with amyotrophic lateral sclerosis The Lancet Neurology, Volume 6, Issue 10, October 2007, Pages 913-925
Sclérose latérale amyotrophique
VENTILATION
NON-INVASIVE INVASIVE
trachéotomie
nocturne
min 4H/jour 24H/24
> 24H/24
- masque facial
- masque nasal
- embout buccal
Sclérose latérale amyotrophique
GASTROSTOMIE
• Si perte de poids 10 % tble déglutition, fatigue
• voie d’administration médicaments : confort et facilité
• qualité de vie améliorée : patient & famille
• amélioration de la survieSclérose latérale amyotrophique
La sclérose latérale amyotrophique :
Physiopathologie
et piste pour de nouveaux traitementsSclérose latérale amyotrophique
Biologie : aggrégation de protéines, inclusions cytosoliques
Métabolisme anormal des neurofilaments
Dysfonction des mitochondries
Perturbation de facteurs de croissance (VEGF)
Altération oxido-redox / neuro-inflammation
Excitotoxicité : défaut de transport du glutamate
Sporadique (90%) or héréditaire (10%)35
Superoxide dismutase (SOD1)
Rosen DR, Siddique T, Patterson D et al. Mutations in Cu/Zn superoxide dismutase
gene are associated with familial amyotrophic lateral sclerosis. Nature 1993; 362:
59-62.
Survival times for SOD1 mutations
G37R, G41D, H46R, and E100K >17 years
G93C ~10 years
The mutation G93A causes a gain-of- E100G and G85R ~5-6 years
L38V, H43R, and G93A ~1-3 years
function : excessive accumulation of
A4V ~1 year
H2O2 and production of other toxic
reactive oxygen species (OH•)
Amyotrophic Lateral Sclerosis
Superoxide dismutase (SOD1)
Rosen DR, Siddique T, Patterson D et al. Mutations in Cu/Zn superoxide
dismutase gene are associated with familial amyotrophic lateral
sclerosis. Nature 1993; 362: 59-62.
Transgenic rat strain
overexpressing the ALS
associated mutated form
of superoxide dismutase
(hSOD1G93A)
Loss of
extension
reflexPerte de poids Score moteur
Lignée de rat
transgéniques
surexprimant la forme
mutée humaine de la SOD1
(hSOD1G93A)
Perte du
réflexe
d’extension
La mort des motoneurones résulte d’une toxicité de la forme
mutée de la SOD1 et non pas d’une altération de la fonction
de l’enzyme elle-mêmeProgression de la pathologie au niveaux moléculaire et cellulaire dans
des souris transgéniques exprimant la protéine SOD1G93A.
Le stade ‘‘symptomatique’’ est celui qui suit le déclenchement de la
perte de poids et l’apparition des faiblesses musculaires (80–100 jours).
« The stars [neurons] could not put on a play alone, and the
more complex the performance, the more support is required »
« Les vedettes [neurones] ne peuvent assumer le spectacle
seules et plus la performance est complexe, plus le soutien des
coulisses est important »Les coulisses du système nerveux
Oligodendrocytes
Astrocytes (& cellules de Schwann) microglie
Modulation de l’environnement
des neurones
Développement Régénération
Physiologie Pathologie
Dans les coulisses du cerveau : les astrocytes!
Neurone
Astrocyte
Rôle dans le
contrôle de
l’excitotoxicité
Vaisseaux Rôle dans le
sanguins contrôle du
métabolismeRôles physiologiques des astrocytes
(non-exhaustive)
• Migration neuronal
• Support physique
• Support métabolique
• Support trophique
• Détoxification de l’ammoniac
• Homéostasis ionique & pH
• Homeostasis des neurotransmetteurs
• Neurogénèse chez l’adulte
• Neuroinflammation
VOLUME 10 NUMBER 5 MAY 2007 NATURE NEUROSCIENCEMotor neuron death in ALS is not cell autonomous
Summary of findings from studies using transgenic mouse models with tissue-specific mutant
SOD1 expression or Cre–Lox/siRNA knockdown of ubiquitously expressed mutant SOD1.
Motor neuron death in ALS is not cell autonomous
Summary of findings from studies using transgenic mouse models with tissue-specific mutant
SOD1 expression or Cre–Lox/siRNA knockdown of ubiquitously expressed mutant SOD1.
X
X X X
XMotor neuron death in ALS is not cell autonomous Summary of findings from studies using transgenic mouse models with tissue-specific mutant SOD1 expression or Cre–Lox/siRNA knockdown of ubiquitously expressed mutant SOD1.
NEJM Volume 326:1464-1468 May 28, 1992
Decreased glutamate transport by the brain and spinal cord in
amyotrophic lateral sclerosis
JD Rothstein, LJ Martin, and RW Kuncl
Abstract
BACKGROUND. Amyotrophic lateral sclerosis (ALS) is a chronic degenerative neurologic disorder characterized by the death of
motor neurons in the cerebral cortex and spinal cord. Recent studies have suggested that the metabolism of glutamate, a
potentially neurotoxic amino acid, is abnormal in patients with ALS. We hypothesized that the high-affinity glutamate transporter
is the site of the defect. METHODS. We measured high-affinity, sodium-dependent glutamate transport in synaptosomes from
neural tissue obtained from 13 patients with ALS, 17 patients with no neurologic disease, and 27 patients with other neuro-
degenerative diseases (Alzheimer's disease in 15 patients and Huntington's disease in 12 patients). The groups were comparable
with respect to age and the interval between death and autopsy. Synaptosomes were prepared from spinal cord, motor cortex,
sensory cortex, visual cortex, striatum, and hippocampus. We also measured sodium-dependent transport of gamma-
aminobutyric acid and phenylalanine in the synaptosomal preparations. RESULTS. In patients with ALS, there was a marked
decrease in the maximal velocity of transport for high-affinity glutamate uptake in synaptosomes from spinal cord (-59
percent, P less than 0.001), motor cortex (-70 percent, P less than 0.001), and somatosensory cortex (-39 percent, P less than
0.05), but not in those from visual cortex, striatum, or hippocampus. The affinity of the transporter for glutamate was not
altered. No abnormalities in glutamate transport were found in synaptosomes from patients with other chronic
neurodegenerative disorders. The transport of gamma-aminobutyric acid and phenylalanine was normal in patients with ALS.
CONCLUSIONS. ALS is associated with a defect in high-affinity glutamate transport that has disease, region, and chemical
specificity. Defects in the clearance of extracellular glutamate because of a faulty transporter could lead to neurotoxic levels of
extracellular glutamate and thus be pathogenic in ALS.
Glutamate
Neurotransmission
Mémoire Excitotoxicité
Apprentissage Plasticité
neuronale
Libération
Activation de récepteurs
Recapture par des transporteursLe glutamate en pathologie
…ou l’intérêt de thérapeutiques anti-glutamatergiques
- Epilepsie :
hyperactivité excitatrices locales (focales)
- Troubles vasculaires cérébraux (‘stroke’) :
ischémie, hémorragie, lésions physiques, …
= perturbation de la balance libération/recapture
- Maladies dégénératives
Chorée de Huntington, Maladie d’Alzheimer, Maladie de Parkinson,
Sclérose latérale amyotrophique
(perturbation de l’homéostasie glutamatergique : cause initiale ou
aggravation?)
Transporteurs du glutamate
Le glutamate dans le SNC
Homogénat de tissu : 10 mM
Fente synaptique : 1 µM
Vésicules synaptiques : 20 mM
GLT-1 : Glutamate transporter 1
GLAST : Glutamate aspartate transporterAnnals of Neurology 38 (1), 73–84, July 1995
Selective loss of glial glutamate transporter GLT-1 in
amyotrophic lateral sclerosis
Jeffrey D. Rothstein, Marleen Van Kammen, Allan I. Levey, Lee
J. Martin, Ralph W. Kuncl
Abstract
The pathogenesis of sporadic amyotrophic lateral sclerosis (ALS) is unknown, but defects in synaptosomal high-affinity glutamate
transport have been observed. In experimental models, chronic loss of glutamate transport can produce a loss of motor neurons
and, therefore, could contribute to the disease. With the recent cloning of three glutamate transporters, i.e., EAAC1, GLT-1, and
GLAST, it has become possible to determine if the loss of glutamate transport in ALS is subtype specific. We developed C-
terminal, antioligopeptide antibodies that were specific for each glutamate transporter. EAAC1 is selective for neurons, while
GLT-1 and GLAST are selective for astroglia. Tissue from various brain regions of ALS patients and controls were examined by
immunoblot or immunocytochemical methods for each transporter subtype. All tissue was matched for age and postmortem
delay. GLT-1 immunoreactive protein was severely decreased in ALS, both in motor cortex (71% decrease compared with control)
and in spinal cord. In approximately a quarter of the ALS motor cortex specimens, the loss of GLT-1 protein (90% decrease from
control) was dramatic. By contrast, there was only a modest loss (20% decrease from control) of immunoreactive protein EAAC1
in ALS motor cortex, and there was no appreciable change in GLAST. The minor loss of EAAC1 could be secondary to loss of
cortical motor neurons. As a comparison, glial fibrillary acidic protein, which is selectively localized to astroglia, was not changed
in ALS motor cortex. Because there is no loss of astroglia in ALS, the dramatic abnormalities in GLT-1 could reflect a primary
defect in GLT-1 protein, a secondary loss due to down regulation, or other toxic processes.
The transgenic rat strain hSOD1G93A
Howland et al., 2002.
PNAS 99, 1604-1609
Focal loss of the glutamate
transporter GLT-1 (EAAT2) in
a transgenic rat model of SOD1
mutant-mediated amyotrophic
lateral sclerosis (ALS).
ExcitotoxicityNature 433, 73 (2005)
57
Lancet Neurology, 2011Ceftriaxone et ALS
• Phase III clinical trial with ceftriaxone in ALS.
NIH and NEALS were sponsors
60 study sites USA, Canada, 2007
8 aout 2012 : stop
August 8, 2012
In July 2012, the DSMB for the NINDS-sponsored clinical trial of ceftriaxone in ALS
recommended that based on existing data the trial be stopped because the study was
unlikely to reach the pre-determined efficacy criteria. The NINDS leadership
concurred. Pre-clinical research identified ceftriaxone as a promising treatment for ALS
therefore it was important for people with ALS to find out if the drug could be beneficial in
ameliorating the disease. The study used a novel seamless adaptive design. Final analysis
and presentation of the results will occur after completion of site monitoring and database
lock. The important contributions of patients, their families and the hard work of the
investigators and their teams made it possible to implement the trial. While all had hoped for
a more positive result, the trial has moved ALS research forward.
Thérapie
génique?
Réprimer
l’expression
de certains
gènes.• Seulement 5% des patients SLA ont une mutation de la SOD1 • Mais les mécanismes pathogéniques semblent tous converger vers une toxicité de la SOD1 (même les cas sporadiques)
La sclérose latérale amyotrophique :
et les déceptions dans la recherche
de nouveaux traitements
ALS après le riluzole :
• Essais cliniques; riluzole combiné à
SR57746A, xaliproden, (1997-99/1999-01)
effet neurotrophique, SOD-Mousemodel+
2 études multicentriques : négatif
ONO-2506PO, cereact, (2001-05/2006-09)
amélioration du métabolisme astrocytaire, SOD-Mousemodel+
2 études multicentriques : négatifTalampanel
Perampanel*
Becampanel
Talampanel (LY300164) : AMPA glutamate receptor
antagonist
Clinical trial in ALS, May 2010 : negative results.
Perampanel in Epilepsy
Clinical phase II, 4 to 12 mg/d efficient in refractory partial
onset seizures, safe and well tolerated.
Clinical trials (3 phase III) positive results in epilepsy
• Memantine (2010)
• Topiramate (2003)
• Lamotrigine (2003)
• Gabapentine (2001)
résultats négatifs dans la SLA.Minocycline
Minocycline :
Antibiotique ,
Anti-apoptotique, anti-inflammatoire
Prolonge la survie des modèles animaux transgéniques
Etude clinique phase III contrôlée:
détérioration plus rapide avec minocycline
tendance à une surmortalité dans le groupe traité
P.Gordon et al. Lancet Neurology 2007
Lithium, PNAS 2008Lithium in the ALS mouse model (PNAS,2008)
“Lithium has neuroprotective effects in cell and animal
models of amyotrophic lateral sclerosis”
“We found no evidence of benefit of lithium on survival in
patients with ALS, but nor were there safety concerns”Nature Medicine, dec. 2011
Nature Medicine, dec. 2011 des premiers essais encourageants Dexpramipexole dans la SLA, Essai clinique phase III (« Empower »). Multicentrique, contrôlé, 900 patients, 8 sous-groupes. Tous les patients reçoivent le riluzole en association. Débute en janvier 2011, recrutement très rapide.
Dexpramipexole, mail du 28/1/2013
• Dear Prof. Maloteaux,
•
• I hope you are doing well!
• Biogen Idec and the Empower team started this year with sad news: the negative results for the
Empower Study (dexpramipexole).
• All activities around this molecule stopped after even looking at the results of another 8 subgroups.
• Biogen Idec will continue to do clinical research in ALS. As soon as I have more details on future
programs I will let you know.
• Best regards,
•
• Marleen
•
• Biogen Idec
• Van Kerckhoven Marleen
• Clinical Research Country Lead Belux
•
• Tel: 00 32 (0)2 711 05 18
• Fax: 00 32 (0)2 219 23 58
• Mobile: 00 32 (0)477 477 001
Dexpramipexole; ALS trial conclusions
• Biogen Idec has announced that the experimental ALS drug has not shown
efficacy in a large-scale trial; its development will be discontinued
• Dexpramipexole, an experimental drug for amyotrophic lateral sclerosis
(ALS), had shown encouraging results in a phase 2 trial but did not
continue to show promise in a phase 3 trial.
• Biogen Idec will discontinue its dexpramipexole development program;
the company has provided phone numbers for people who want more
information.Essais cliniques dans la SLA ;
autres pistes
• Ozanezumab, GSK1223249, inhibiteur Nogo-A, augmente
croissance neuritique, ALSM+, phase IIa
• Gilenya, fingolimod, inhibition lymphocytaire, ALSM+, phase IIa
• Arimoclomol, BRX-220, stimule des protéines chaperon et la
réparation protéique, ALSM+, phase II
• E0302, mecobalamine, phase III (Japon)
• CK-2017357, Tirasemtiv® activateur de la troponine
musculaire, augmente la sensibilité au Ca++ dans les muscles,
ALS-SODmodel +, phase II
Essais cliniques dans la SLA ;
autres pistes
• P7C3, aminopropyl carbazoles, neuroprotection ALSmodel+, préclinique.
• TRO19622, olesoxime, neurotrophique et protecteur mitochondrial, ALSmodel+,
essai ALS (-), phase II dans l’amyotrophie spinale
• NP001 (bleu de méthylène), protecteur mitochondrial, inhibe NO et microglie,
phase III
• Tamoxifen et creatine, effet neuropretecteur et périphérique (CK), ALSmodel+,
phase II
• Mexiletin, antiarythmique, bloquer sodique, anti-crampes, phase II
• Facteurs de croissance: BDNF (-), VEGF ALSModel+, phase II ip
• Antisense contre SOD1, phase I ic (NCT01041222), forme SOD1fam.
• Vaccination (prot. SOD mutante), immunisation par Ac anti-SOD1: préclinique
MouseModel+.
• Cellules souches transplantées…Quelques remarques finales : • Pourquoi les animaux transgéniques ne sont-ils pas prédictifs ? • L’utilisation prématurée (off-label) ne doit pas être encouragée (lithium, ceftriaxone, minocycline !) • Le riluzole dans les conditions et critères d’essais cliniques actuels serait-il efficace ? • Des marqueurs (« phase 0 ») de la SLA, maladie hétérogène, permettraient-ils de sélectionner des patients répondeurs ?
Vous pouvez aussi lire

















































