Puces à ADN (DNA microarrays)
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous

Puces à ADN (DNA microarrays) Sébastien Lemieux (s.lemieux@umontreal.ca) Laboratoire de bioinformatique structurale et fonctionnelle Institut de Recherche en Immunologie et Cancer - IRIC /" Département d'Informatique et de Recherche Opérationnelle - DIRO Université de Montréal
Plan • Survol d'une expérience typique • Allison et al. Microarray data analysis: from disarray to consolidation and consensus. Nat. Rev. Genet. (2006) 7(1):55-65 • Ressources disponibles (bases de données, logiciels) • Variations • Séquençage haut-débit
Transcriptomique
Puce à ADN
Déroulement d'une expérience d'expression
• L'impression: attacher des brins d'ADN
(sondes) sur une matrice solide.
• On peut commercialement se procurer des puces
pré-imprimées avec les sondes de notre choix
(Affymetrix, Agilent, Nimblegen).
• Le marquage: coupler de manière covalente
les ARNm avec un fluorochrome (rouge/vert?).
• Soit les sondes sont conçues pour être
complémentaires aux ARNm,
• soit on fait une rétrotranscription des ARN en ADNc
avant le marquage.
• L'hybridation: on laisse s'apparier de manière
spécifique les ARNm (ou ADNc) aux sondes.
• Acquisition de l'image avec un scanner laser.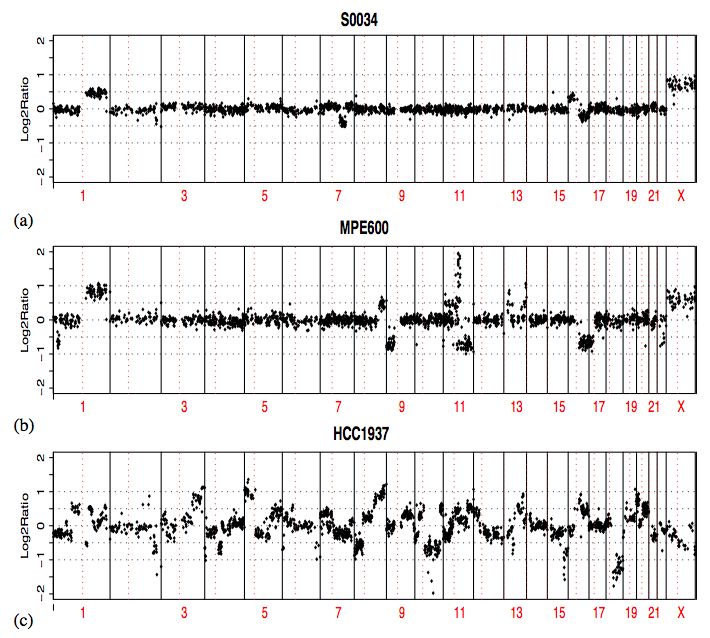
Déroulement d'une expérience d'expression • Acquisition de l'image avec un scanner laser. • Griding / quantification: convertir l'image en niveaux d'intensité par sonde. Cette intensité est proportionnelle à la concentration de ARNm correspondant. • Normalisation: retirer un biais au niveau des log-ratios qui dépend de l'intensité. • Sommarisation: combinaison des niveaux de plusieurs sondes. • Inférence: identifier les gènes régulés. • Analyse de groupe: identifier des groupes de gènes régulés.
Concepts de base
• Marquage (dye, fluor): Cy3/Cy5 ou Alexa.
• Avantage et biais liés à la rétrotranscription.
• Le marquage direct permet d'hybrider avec
plusieurs "couleurs" une même puce.
• Sonde (probe): oligonucléotides (spotted,
in situ) ou cDNA.
• La longueur de la sonde détermine la
spécificité de l'hybridation.
• Le design d'une puce consiste à choisir les
séquences des sondes.
• Réplication: biologique ou technique.
• Intensités, ratios ou log-ratios?
Deux types de procédés de fabrication
www.affymetrix.com
cDNA ou"
Oligonucléotides seulement
oligonucléotides
Types de puces
• cDNA:
• Des cDNA sont amplifiés, puis liés de manière covalente au substrat de la puce.
• L'ensemble du gène est disponible pour déterminer la spécificité.
• Peut-être fait en l'absence de la séquence du génome.
• Très peu utilisés.
• Oligonucléotides:
• Technologie dominante
• Affymetrix (25 nt): photolithographie, chimie en phase solide.
• Agilent (25-60 nt, 244k sondes / puce): technologie similaire aux imprimantes à jet
d'encre.
• Nimblegen (50-75 nt, 2.1M sondes / puce): procédé photochimique utilisant un
masque électronique (DLP)
• Spotted arrays: la synthèse des oligos est faite de manière conventionnelle puis
déposée sur la puces.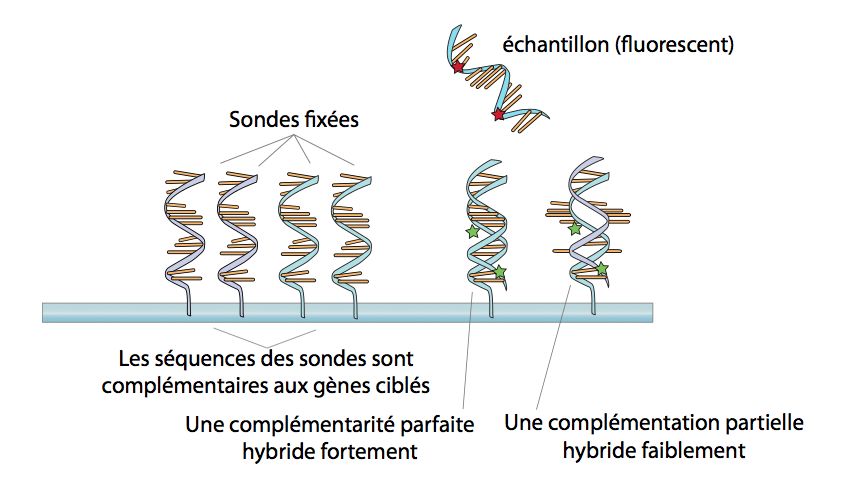
GeneChips - Affymetrix
• Plateforme la plus courante, très grande
densité (5 millions de sondes?).
• Perfect Match (PM) vs. Mismatch (MM):
• Initialement, visait à distinguer la contribution à
l'intensité d'hybridations non-spécifiques. La
technique la plus simple consiste à utiliser PM -
MM.
• De plus en plus de groupes suggèrent d'ignorer les
lectures MM.
• Nécessite un appareillage spécialisé pour
l'hybridation et la digitalisation.
• Une seule "couleur" (marquage indirect).
• Haute reproductibilité au niveau de la puce.
• Ne se traduit pas toujours par une haute
reproductibilité au niveau de la biologie observée.Allison et al. Microarray data analysis: from disarray to consolidation and consensus. Nat. Rev. Genet., 7(1):55-65, 2006."
Design expérimental
• La réplication biologique est essentielle.
• Dans tous les cas, la réplication biologique est préférable à une réplication
technique.
• Sauf dans les cas de développement de la technologie, les réplicats techniques
n'apportent aucune information.
• Le dye-swap devrait être combiné à un réplicat biologique.
• Calcul de puissance: 5 réplicats sont recommandés...
• Peu respecté en pratique, question de coût. Un nombre inférieur de réplicats est
souvent adéquat pour une étude exploratoire.
• La méthode d'inférence utilisée nous permet d'ajuster le compromis sensibilité vs.
spécificité.Design expérimental
• Le pooling d'échantillons biologiques peut être utile.
• Compromis entre réplicats biologiques et "pooling". Estimer la variance vs. réduire
la variance.
• Approche assez peu utilisée, a reçu mauvaise presse par le passé dû à
l'impossibilité d'estimer la variance.
• Éliminer du design les facteurs qui ne sont pas à l'étude.
• Biais vs. variance. On peut réduire la variance par les réplicats, ou en tenir compte
dans l'inférence. Mais les biais expérimentaux généreront nécessairement des faux
positifs impossibles à détecter.
• Lorsqu'un tel design est impossible, on tente d'"orthogonaliser" ces facteurs.Design expérimental
www.transcriptome.ens.fr/sgdb/contact/downloa/200601_Lelandais1.pdf
Nécessaire normalisation
Allison et al. Nat. Rev. Genet., 7(1):55-65, 2006.Preprocessing
• Aucun consensus sur la méthode de sommarisation et normalisation
idéale à utiliser.
• Utilisation dans tous les cas d'une transformation log. Soit sur les intensités (alors
on s'intéresse aux différences d'intensité), soit sur les ratios d'intensités.
• Dans la pratique, on utilise une normalisation loess suivie d'une transformation log
pour des puces 2 couleurs. Pour les puces Affymetrix (une couleur), on préfère les
algorithmes MAS5, RMA ou GCRMA. Ces méthodes retournent des valeurs sur une
échelle log.
• Pas de consensus sur la façon d'évaluer la qualité d'une expérience.
• Difficulté d'obtenir un échantillon de référence.
• La "qualité" d'une expérience varie en suivant beaucoup de facteurs imprédictibles.
• Le critère de base que serait la reproductibilité est difficile à évaluer puisqu'il varie en
fonction des sondes et des conditions expérimentales. Le coût des réplicats
techniques est un obstacle important.Inférence statistique
• Objectif: Identifier les gènes régulés entre différentes conditions.
• Variations: ChIP-chip, multiples conditions, CGH, tiling arrays, etc.
• Erreurs de type 1: faux positifs
• Erreurs de type 2: faux négatifs
• L'utilisation d'un seuil sur le ratio (fold-change) n'est pas suffisante.
• Ne tient pas compte de la variabilité des observations.
• Ne tient pas compte du nombre de réplicats.
• Une approche basée sur le ratio correspond à un test avec régularisation extrême.
• Dans tous les cas, si une opération de moyenne ou de calcul d'écart-type est fait,
elle doit l'être sur des valeurs en échelle log."
Ne jamais faire la moyenne des valeurs d'intensité non-transformée."
Ne jamais faire la moyenne de ratios observés dans différents réplicats.Inférence statistique
• L'utilisation d'un terme de régularisation est souhaitable.
• L'estimation de la variance est imprécise.
• L'idée est de baser cet estimé sur des informations a priori ou acquise à partir
d'autres gènes.
• Aucun consensus n'existe quant au choix de la meilleure méthode de régularisation.
Les méthodes limma-ebayes (bioconductor), SAM, cyber-T, etc. présentent toutes
un terme de régularisation.Inférence statistique
• Tester les gènes en groupe permet de minimiser les erreurs d'estimation
dues au faible nombre de réplicats.
• Analyses par Gene Ontology ou autre classification. Ex: GSEA.
• Permet aussi d'obtenir des observations de plus haut niveau. Qui sont par contre
difficiles à valider expérimentalement...
• Excellente revue des méthodes disponibles: Rivals et al. Enrichment or depletion of
a GO category within a class of genes: which test? Bioinformatics, 23:401-7, 2007.
• La correction pour tests multiples est nécessaire.
• La méthode de False Discovery Rate (FDR) est généralement recommandée.
• Fait à noter: ces corrections ont pour but de contrôler le taux de faux positifs (fixer
une borne supérieure) mais aucun ne donne de garantie quant à la sous-estimation
du taux de faux positifs. La forte corrélation de l'expression des gènes aggrave ce
problème.
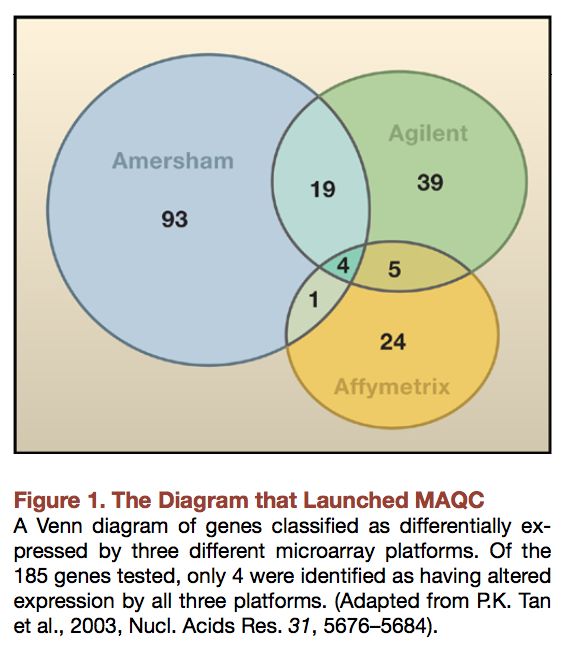
• Un mot sur les diagrammes de Venn...Diagrammes de Venn
• Très utilisée pour représenter les
intersections entre listes de gènes
régulés.
• Simple et facile à comprendre.
• Devient graphiquement très complexe à
partir de 4 ensembles.
• Grande influence des seuils choisis
pour chaque ensemble.
• En grande partie responsable de la
mauvaise presse faite aux puces à
ADN.
• Pas de consensus clair sur
l'approche à utiliser pour identifier
l'intersection (ou la différence) entre
plusieurs expériences.Classification
• Objectif: identifier des groupes d'échantillons ou de gènes avec des
profiles d'expression similaires.
• Les échantillons groupés indiquent un mécanisme similaire.
• Les gènes groupés indiquent une co-régulation à travers les conditions analysées.
• La classification non-supervisée n'est pas toujours appropriée:
• Algorithmes courants: K-mean, Self-Organizing Maps (SOM), hierarchical
clustering. À la limite: Principal Component Analysis, Singular Value
Decomposition.
• Devrait uniquement servir d'analyse exploratoire lorsqu'aucune catégorisation n'est
connue sur les échantillons.
• Tout type de classification doit inclure une validation:
• Non-supervisée: bootstrap.
• Supervisée: k-fold cross-validation, leave-one-out.Classification
• On obtient toujours des groupes:
• exemple de double hierarchical
clustering;
• données aléatoires, triées par leur
différence moyenne entre
"échantillons" 1-5 et 6-10.
• Les 200 "gènes" avec les
différences les plus grandes sont
présentées.Validation expérimentale
• Objectif: confirmer les observations faites par l'expérience de puce.
• Méthodes utilisées:
• qRT-PCR: amplification spécifique par PCR avec une sonde centrale couplée à un
fluorophore. La courbe d'amplification indique avec précision la concentration
initiale. Spécifique (3 sondes). Coûteux. (Taux de confirmation: 50-80%)
• Northern Blot: les ARN sont séparés par longueur sur un gel d'agarose puis une
sonde (couplée avec un isotope radioactif) est ajoutée pour identifier et quantifier
l'ARNm d'intérêt. Approche surtout qualitative, aucun appareil spécialisé.
• Quels gènes valider?
• En pratique, on valide les gènes pour lesquels on continue le travail ou sur lesquels
nos conclusions se basent.
• Corrélations entre qRT-PCR et puces à ADN: les niveaux de régulation
observés sont plus élevés en PCR qu'en puce...Base de données publiques
• Base de donnée générale:
• Gene Expression Omnibus - Database of gene expression data from NCBI.
• ArrayExpress - Database of gene expression and other microarray data at the EBI.
• Stanford Microarray Database - Data from microarray experiments, and the
corresponding image files.
• Par tissus, commerciales ou meta-analyses:
• Allen Brain Atlas - Database of gene expression patterns in the mouse brain.
• Gene Expression in Tooth - Expression of different genes in the developing tooth.
• GeneSifter Data Center - GeneSifter microarray data analysis system in selected
conditions.
• Genevestigator - Multi-organism microarray database and expression meta-analysis
tool.
• BioGPS - Database on gene function and structure, from Novartis Research
Foundation.BioConductor et R
• R: www.r-project.org
• Environnement de programmation statistique open source.
• Apprentissage difficile, très grande flexibilité.
• Langage de programmation très lent, pas idéal pour de grands projets nécessitant
des analyses rapides.
• Bioconductor: www.bioconductor.org
• Ensemble de librairie R facilitant l'analyse de données de puces à ADN.
• Le code source de tous les algorithmes sont disponible.
• Environnement privilégié par une majorité de développeurs de méthodes
statistiques.
• Facilité à automatiser des analyses complexes et non-conventionnelle.
• Bioinformatics and Computational Biology Solutions Using R and
Bioconductor, R. Gentleman et al., Springer, 2005.Variations - CGH
• Comparative Genomic Hybridization:
• On fragmente, puis marque l'ADN génomique.
• L'hybridation est faite sur une puce de tiling (des
sondes sont également distribuées sur toute la
séquence génomique).
• Une variation d'intensité indique une
modification du nombre de copies de la
séquence correspondant à la sonde.
• La densité des sondes sur le génome
détermine la taille minimale des aberrations
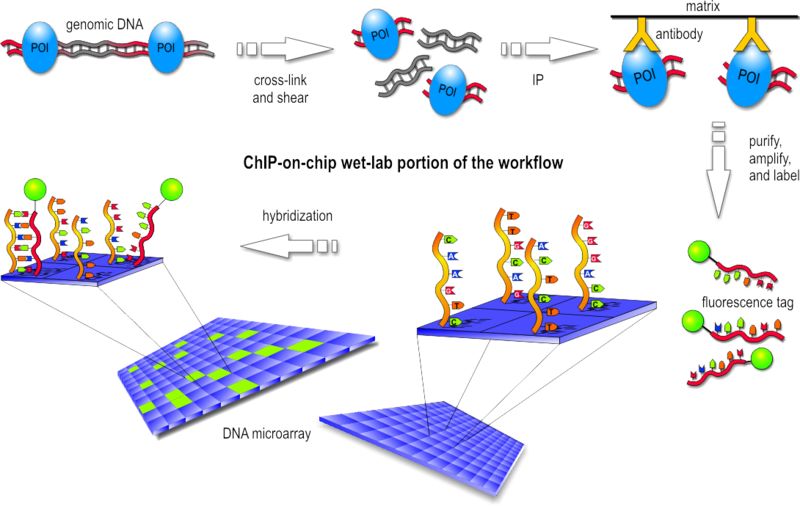
qui peuvent être détectées.Variation - ChIP-on-chip
• Chromatin-ImmunoPrecipitation on
chip:
• On construit d'abord une souche dans
laquelle un facteur de transcription est
combiné à un tag moléculaire (TAP,
HA).
• Par traitement chimique, on lie de
manière covalente les protéines à
l'ADN génomique. L'ADN génomique
est ensuite fragmenté et filtré pour
retirer les brins liés au tag.
• Les protéines sont retirées, l'ADN
marqué puis hybridé sur une puce de
tiling.
• Indique les positions dans le
génome où un facteur de
transcription se lie.Variation - ChIP-on-chip
Séquençage haut-débit (next-gen)
• Une technologie pouvant remplacer les puces à ADN...
2011:
150-200 Gb / 4 jours (Illumina HiSeq 2000"
80-120 Gb / 4 jours (SOLiD 5500xl)Séquençage haut-débit
• Approche expérimentale
• Un grand nombre (300-500 x 106) de courts fragments (50-100 nt) sont séquencés
• Comparatif du coût par base (0.30$ par million de bases séquencées)
• Mapping
• Consiste à aligner chaque fragment avec un génome de référence
• L'algorithme classique pour accomplir ce type de tâche est BLAST: lorsqu'on
considère la taille du jeu de données, on constate que BLAST est trop lent (50-200
jours de temps CPU pour aligner ce qu'un séquenceur produit en 4 jours)
• Le débit des séquenceurs augmente beaucoup plus rapidement que la puissance
des processeurs
• Assemblage de novo
• Représente encore un défi important
• Exemple: Simpson et al., ABySS: A parallel assembler for short read sequence data,
Genome Research, 19:1117-23, 2009.ABI SOLiD - Next-generation sequencing • Plusieurs technologies sont disponibles (Illumina, PacBio, Helicos, ...) • Le principe commun à toutes ces approches est le séquençage en parallèle de millions de fragments attachés à un support solide.
Acquisition des données • Pour chaque position: • 4 images seront prises par un système de caméra automatisée • la couleur indique la base présente à cette position pour le fragment lié à la bille • Les images ne sont typiquement pas conservées: ~1 TB d'image pour environ 50 GB de séquence.
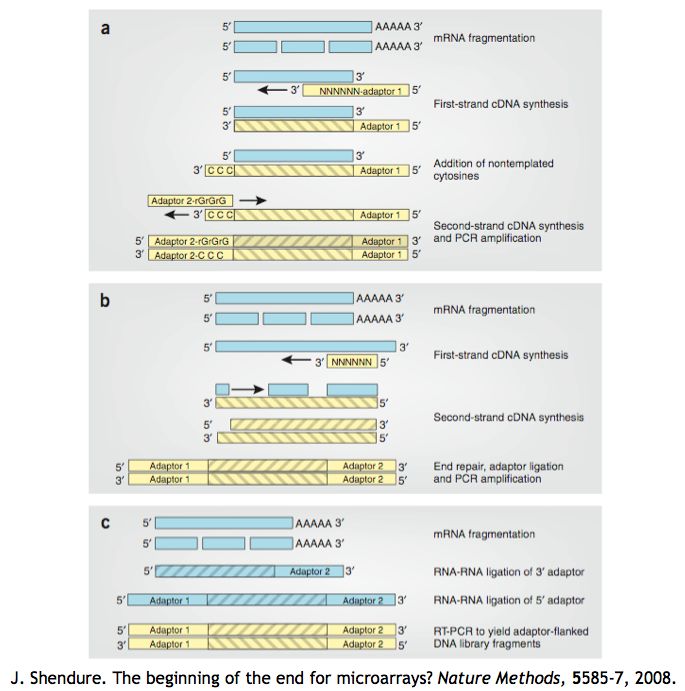
RNA-seq - Construction de la librairie
Séquençage: ADN vs. ARN
Alignement - Spaced seeds
Choi et al., Good spaced seeds for homology
search, Bioinformatics, 20(7):1053–9, 2004.Alignement - Indexes FM (BW transforms) • Bowtie: Langmead et al., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome, Genome Biology, 10:R25, 2009. (50 x 106 reads per hour per CPU) Transformation de Burrows-Wheeler
Identification de variants
• On comptabilise le nombre de fragments indiquant la
base consensus et la base variante.
• Si on compare deux échantillons, un test exact de Fisher
est souvent utilisé:
A autre
Échantillon #1 124 113
Échantillon #2 2 87
• Que faire si un seul échantillon est analysé?Fragments en paires (mated short reads)
• Chaque fragment sera séquencé deux fois,
résultant en 2 séquences de 50-75 nt
• La séparation dépend de la taille moyenne
des fragments sélectionnés
www.illumina.comFragments en paires (mated short reads) • Exemple: Medvedev et al., Detecting copy number variation with mated short reads, Genome Research, 20:1613-22, 2010.
Votre mission, si vous l'acceptez...
Vous pouvez aussi lire

















































