CONCEPTION DE SONDES FISH - Maîtrise de Biochimie
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
UNIVERSITE HENRI POINCARE, NANCY I
FACULTE DES SCIENCES ET TECHNIQUES
Maîtrise de Biochimie
Année universitaire 2003/2004
CONCEPTION DE SONDES FISH
Mémoire présenté par :
DERIVE Marc
STAGE EFFECTUE AU
LABORATOIRE DE CYTOGENETIQUE MOLECULARE
CENTRE HOSPITALIER UNIVERSITAIRE
BRABOIS
1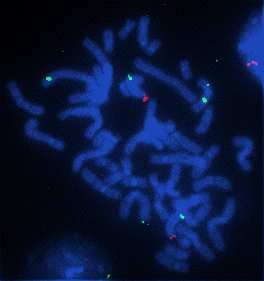
2
Ac : Anticorps
A.D.N. : Acide DéoxyriboNucléotidique
BAC : Bacterial Artificiel Chromosome
BET : Bromure d’Ethidium
CEP : CEntromeric Probes : sondes centromèriques
Cf : Concentration finale
CGH : Hybridation Génomique Comparative
DAPI : 4’-6-diamidino-2-phénylindole
DMSO : Di Méthyl Sulfoxide
dNTP : di-Nucléotide-Tri-Phosphate
DOP-PCR : Degenerate Oligonucléotide Primer – Polymerisation Chain Reaction
E. Coli : Escherichia Coli
F : Formamide
FISH : Hybridation fluorescente in situ
FITC : Isothiocyanate de fluorescéine
H2O mQ : H2O distillée qualité milliQ
kb : kilo bases
LSI : Locus Specific Intern : sondes internes locus spécifiques
Mb : Méga bases
MECP2 : Methyl-CPG-Binding Protein 2
NP-40 : Nonidet P-40
PAC : P1 Artificiel Chromosome
PBS : Phosphate-Buffere Saline.
pdb : paires de bases
QI : Quotient intellectuel
RCA : Rolling Circle Amplification : Amplification en cercle roulant
RPMI : Roswell Park Memorial Institute
SDS : Sodium-Dodécyl-Sulfate
SSC : Saline Sodium Citrate
STS : Séquence Tag Sites : marqueurs positionnels de l’ADN
Tm : Température de fusion (melting)
3
INTRODUCTION................................................................................................................................................. 5
I. LES CHROMOSOMES................................................................................................................................... 6
I.1. Généralités et nomenclature ..................................................................................................................... 6
I.2. Les télomères (schéma 3 en annexe 2)...................................................................................................... 7
II. EVOLUTION DES TECHNIQUES DIAGNOSTIQUES .............................................................................. 8
III. CONCEPTION DE SONDES FISH.............................................................................................................. 9
III.1. La DOP-PCR ......................................................................................................................................... 9
III.2. Le kit d’amplification GenomiPhi ........................................................................................................ 10
III.3. Intérêt ................................................................................................................................................... 10
IV. OBJECTIFS DE CE STAGE ............................................................................................................................... 11
MATERIEL ET METHODES....................................................................................................................... 12
I. LE KIT VYSIS « TOTELVISIONTM MULTI-PROBES PANEL » ........................................................................ 13
I.1. Obtention de métaphases ........................................................................................................................ 13
I.2. Pré-Traitement à la pepsine.................................................................................................................... 13
I.3. Les sondes............................................................................................................................................... 14
I.4. Hybridation des sondes........................................................................................................................... 14
I.4.a. Hybridation ...................................................................................................................................... 14
I.4.b. Rinçage ............................................................................................................................................ 15
I.4.c. Les fluorochromes............................................................................................................................ 15
I.4.d. L’analyseur d’images....................................................................................................................... 15
II. CONCEPTION DE SONDES FISH ............................................................................................................ 16
II.1. Amplification des PACs ......................................................................................................................... 16
II.1.a. Les PACs utilisés ............................................................................................................................ 16
II.1.b. La DOP-PCR .................................................................................................................................. 16
II.1.c. Utilisation du kit GenomiPhi .......................................................................................................... 17
II.2. Marquage des sondes ............................................................................................................................ 18
II.3. Précipitation de l’ADN marqué............................................................................................................. 18
III. HYBRIDATION DES SONDES SUR LAME ............................................................................................ 18
III.1. hybridation ........................................................................................................................................... 18
III.2. Rinçage et détection immunochimique................................................................................................. 19
RESULTATS....................................................................................................................................................... 20
I. LE KIT TOTELVISIONTM MULTI-PROBES PANEL............................................................................................ 21
II. LES SONDES CONÇUES PAR DOP-PCR ET PAR LE KIT GENOMIPHI ................................................................. 21
II.1.Amplification d’ADN par DOP-PCR...................................................................................................... 21
II.1.a. Les produits de la première PCR .................................................................................................... 21
II.1.b. Les produits de deuxième PCR....................................................................................................... 22
II.2. Amplification d’ADN par le kit Genomiphi ........................................................................................... 23
II.3. Marquage des produits d’amplification ................................................................................................ 23
II.4. Hybridation ........................................................................................................................................... 23
DISCUSSION ...................................................................................................................................................... 24
I. LE KIT TOTELVISIONTM MULTI-PROBES PANEL ............................................................................................... 25
I.1. Les sondes............................................................................................................................................... 25
I.2. les étapes de rinçage............................................................................................................................... 25
II. LES SONDES CONÇUES PAR DOP-PCR........................................................................................................... 26
II.1. Produits de la DOP-PCR ...................................................................................................................... 26
II.2. La deuxième PCR .................................................................................................................................. 26
III. LES SONDES CONÇUES PAR LE KIT GENOMIPHI ............................................................................................. 27
CONCLUSIONS ET PERSPECTIVES ............................................................................................................ 28
BIBLIOGRAPHIE.............................................................................................................................................. 30
ANNEXES………………………………………………………………………………………………………..33
4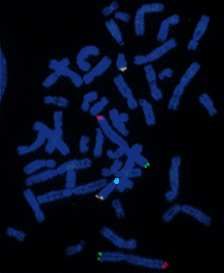
INTRODUCTION
5
L’ADN est le support de l’information génétique. Il est contenu dans le noyau de
toutes les cellules de notre organisme. Cet ADN est structuré en chromosomes lors de la
division cellulaire, au stade de la métaphase. Ils sont classés selon leur morphologie. Dans
l’espèce humaine, 23 paires de chromosomes définissent le caryotype normal, dont deux
chromosomes sexuels X ou Y.
Cependant, certaines anomalies peuvent survenir au cours des divisions cellulaires,
notamment pendant la gamétogenèse, ou encore pendant la formation de l’embryon. De
nombreuses anomalies ne sont pas viables et l’embryon meurt in utero, c’est l’avortement
spontané. Mais d’autres permettent tout de même le développement d’un embryon.
Certaines de ces anomalies sont connues, comme la trisomie 21, résultant de la
fécondation d’un gamète normal et d’un deuxième comportant 2 chromosomes 21.
Outre les anomalies de nombre des chromosomes, les anomalies de structure sont
responsables de déficiences mentales (QIpar le numéro du chromosome, la lettre du bras impliqué, le numéro de la bande et
éventuellement de la sous-bande. Par exemple, la région correspondant à l’astérisque sur le
schéma 2 en annexe1 est notée 21q22.3.
I.2. Les télomères (schéma 3 en annexe 2)
L’extrémité de tous les chromosomes eucaryotiques est composée de régions riches en
nucléotides T et G. Chez les vertébrés, on observe des répétitions de (TTAGGG)n sur 2 à 15
kb. Ils sont indispensables à la réplication complète de l’ADN et subissent au cours de ce
processus une dégradation exonucléolytique, une des causes importantes du vieillissement
cellulaire. Ces successions de séquences (TTAGGG)n sont immédiatement précédées de
répétitions d’ADN, sur quelques centaines de kilobases, dont le rôle est inconnu.
En amont de ces séquences est représenté la région subtélomèrique. D’après certaines
études, [FLINT et al. 1997], [KNIGHTS and FLINT, 2003], elle est divisée en deux sous-
régions, l’une distale et l’autre proximale, espacées par des répétitions (TTAGGG)n
dégénérées. Ces dernières serviraient à compartimenter les domaines subtélomèriques dans le
noyau.
Plusieurs aspects de la structure et de la fonction des télomères suggèrent que les
remaniements cryptiques de ces régions seraient impliqués dans la survenue d’un retard
mental. Au cours de la méiose, l’appariement des chromosomes est initié au niveau des
télomères. Les régions subtélomèriques sont riches en pseudogènes et en séquences répétées
qui partagent des homologies entre chromosomes non homologues, à l’origine d’un
mésappariement au tout début de la prophase, [FLINT et al., 1996 et 1997]. Par exemple, les
subtélomères des bras longs des chromosomes X et Y contiennent le gène du récepteur de
l’interleukine 9. Des copies partielles de ce gène (pseudogènes) sont retrouvées au niveau des
subtélomères des régions 16p, 9p, et 10q avec un haut degré de similarité dans les séquences
[KERMOUNI et al., 1995]. Un taux de recombinaison élevé est observé au niveau de ces
régions, où la concentration en gènes serait la plus élevée [SACCONE et al., 1992], et dont le
déséquilibre causé par des délétions ou des duplications aurait des conséquences
phénotypiques plus importantes que d’autres régions du génome moins riches en gènes.
Les données de la littérature actuelle [JIE XU and ZHONG CHEN, 2003], [KNIGHT
et al.1999], [FLINT AND KNIGHT, 2003] relèvent que 4 à 7% des retards mentaux
idiopathiques seraient dus à une aneusomie subtélomèrique, parfois dues à des réarrangements
complexes [DAVIES et al., 2003]. Parmi les anomalies observées, 50% d’entre elles sont
7héritées d’une translocation parentale équilibrée avec donc un risque de récurrence important
pour la fratrie et éventuellement les autres apparentées.
L’origine de ces réarrangements peut par exemple correspondre au mésappariement et
à l’événement de recombinaison illégitime entre séquences répétées type Alu [KNIGHTS and
FLINT 2003], [FLINT et al., 1996]. Il est difficile d’identifier pour le moment les gènes mis
en jeu, car les délétions connues aujourd’hui sont de quelques Mb en moyenne, d’où le
nombre important de gènes potentiellement concernés. [FLINT AND KNIGHT, 2003].
II. EVOLUTION DES TECHNIQUES DIAGNOSTIQUES
Le seuil de résolution des techniques conventionnelles de cytogénétique ne permet pas
d’identifier des anomalies de petites tailles (inférieures à 5-10 Mb) [KNIGHT et al. 1997] et
surtout sur les régions subtélomèriques.
Ainsi, en 1995, Jonathan Flint a le premier étudié un moyen de diagnostiquer les
réarrangements des régions subtélomèriques dans le cadre des retards mentaux idiopathiques
[FLINT et al. 1995]. La technique FISH (Hybridation Fluorescente in situ) est utilisée pour le
criblage des régions subtélomèriques à l’aide de sondes spécifiques de chacune de ces régions
[KNIGHT et al. 1997]. Ces sondes correspondent à des BACs et des PACs marqués par des
fluorochromes à l’aide de la technique de nick translation. L’originalité de cette approche a
été l’hybridation de l’ensemble de ces sondes (41 sondes marquées par deux fluorochromes :
FITC vert et rhodamine rouge) en une seule expérience sur une lame microscopique
compartimentée en 24 aires, chacune d’entre elles étant spécifique d’une paire
chromosomique.
Cette technique développée en 1997 utilise 24 mélanges de sondes, elle est difficile
d’application [SISMANI et al., 2001] et a donc été simplifiée. Le kit Vysis « ToTelVisionTM
Multi-probes panel » utilise 15 mélanges de combinaisons variables de 62 sondes au total,
sondes subtélomèriques, centromèriques (CEP) ou locus spécifique (LSI).
Son avantage par rapport à la technique utilisée en 1997 consiste en l’utilisation d’un
fluorochrome supplémentaire, l’aqua (bleu aqua), et d’une couleur orange supplémentaire
résultant du mélange des fluorochromes FITC (vert) et Rhodamine (rouge). Les 4
couleurs ainsi obtenues permettent de réduire le nombre de mélanges de sondes à 15.
Chacune de ces sondes couvre un locus d’environ 300 kb.
8J’ai eu l’occasion durant ce stade de me familiariser avec les techniques
d’hybridation in situ en traitant plusieurs dossiers de patients atteints de retards
mentaux à l’aide du kit ToTelVisionTM.
III. CONCEPTION
CONCEPTION DE SONDES FISH
De nombreux progrès en biologie moléculaire appliquée au domaine de la
cytogénétique permettent de faire évoluer la technique FISH. J’ai donc également été
sollicité durant ce stage pour la mise en application de la DOP-PCR (Degenerate
Oligonucleotide Primed -PCR) et du kit d’amplification GenomiPhi (Amersham
Biosciences) à la conception de sondes chromosomiques fluorescentes.
III.1. La DOP-PCR
Le génome humain est compose de séquences répétées comme les séquences Alu,
estimées à 900 000 copies dans le génome humain. La PCR-Alu est une technique
d’amplification globale de l’ADN humain grâce à l’utilisation d’amorces spécifiques de ces
répétitions. Mais ces séquences ne sont pas uniformément reparties dans le génome, elles sont
par exemple relativement rares dans les bandes G [Genome mapping], ce qui crée des « biais
d’amplification » [Telenius et al., 1992].
En 1992, H. Telenius développe la technique de DOP-PCR. Cette technique amplifie
également de manière globale l’ADN cible, sans restrictions dues à sa complexité, ni de
l’espèce dont il est issu et surtout sans en connaître la séquence. Mais la DOP-PCR proposée
est une méthode d’amplification plus homogène de l’ADN, à partir de séquences
hexamèriques reparties de façon homogène dans le génome. Cette technique est utilisée pour
l’amplification et le marquage d’ADN purifié, de chromosomes triés, micro-disséqué, afin
d’obtenir des sondes spécifiques d’un chromosome utilisable pour la FISH ou la CGH
(Hybridation Génomique Comparative) [JONVEAUX et al., 1996], [HIROSE et al., 2001].
La technique a récemment été modifiée pour être plus spécifique du génome humain.
Trois nouvelles amorces ont été conçues, dont la partie 3’ comporte une séquence
hexamèrique répétée spécifiquement dans l’ADN humain [FIELGER et al., 2003]. Ces
amorces amplifient donc préférentiellement l’insert d’un vecteur quand ce dernier est
bactérien (E. Coli) et que l’insert est constitué d’ADN humain.
9III.2. Le kit d’amplification GenomiPhi
Ce kit est commercialisé par Amersham (www.amershambiosciences.com ou
www.genomiphi.com). Il utilise l’ADN polymérase Phi29 du bactériophage du même nom
pour une amplification exponentielle d’ADN simple ou double brin, linéaire on circulaire
(plasmides, BAC, PAC) durant une réaction isothermique (30°C) de 16 heures.
Dans le cas de l’amplification d’ADN circulaire, on parle d’ « amplification en cercle
roulant à multiples amorces » (RCA à multiples amorces). Cette réaction consiste en
l’extension d’amorces hexamèriques, aléatoirement liées à la matrice d’ADN circulaire,
exonucléase résistantes (exo-résistantes ou exo-). En effet, les deux derniers nucléotides en 3’
sont liés par des liaisons triphosphates (5’ – NpNpNpNpppNpppN – 3’), ceci afin d’éviter leur
dégradation par l’activité exonucléase 3’→5’ de l’ADN polymérase Phi29 [DEAN et al.,
2001]. Cette activité correctrice permet une réplication fidèle de l’ADN (le ratio d’erreur est
de 1 pour 106 – 107 bases. (Schéma 4 en annexe 3).
On arrive ainsi à une amplification d’environ 10 000 fois la quantité d’ADN présente
initialement dans le tube, avec le minimum de biais d’amplification [HOSONO et al., 2003].
Les produits de PCR sont double brin, et peuvent donc être utilisées pour différentes
techniques comme le clonage ou le marquage [DEAN et al., 2001].
III.3. Intérêt
De manière générale, la DOP-PCR et le kit GenomiPhi pourraient servir à synthétiser
des sondes FISH simplement et quotidiennement avec le matériel et au sein du laboratoire de
cytogénétique moléculaire de Brabois, sans avoir recours aux lots de sondes commercialisés
par les laboratoires privés.
En effet, la conception de sondes par nick translation nécessite de disposer d’une
grande quantité du vecteur d’intérêt pour le marquer. Les deux méthodes utilisées ici sont
capables d’amplifier de l’ADN initialement présent en petite quantité. Il serait donc
envisageable de réaliser directement des sondes à partir de « mini-prep » de cellules
bactériennes. Sachant que la DOP-PCR est capable d’amplifier spécifiquement l’insert
d’ADN humain contenu dans un vecteur d’origine bactérien (E. Coli), il serait possible
d’extrapoler cette technique et de l’utiliser directement sur un lysat bactérien, sans isoler le
vecteur du reste de l’ADN génomique de la bactérie.
La DOP-PCR et le kit GenomiPhi pourraient donc permettre de synthétiser
simplement les 41 sondes nécessaires au criblage de l’ensemble des régions subtélomèriques
10des chromosomes humains, à partir des BAC et PAC correspondants. Le but serait de réaliser
de nouvelles combinaisons de fluorochromes et donc de synthétiser des sondes de couleurs
plus variées afin de réduire encore le nombre de mélanges de ces dernières.
IV. Objectifs de ce stage
En premier lieu, je me suis familiarisé avec les techniques d’hybridation in situ par le
biais de l’utilisation du kit « ToTelVisionTM Multi-probes panel ».
Puis, mon travail a consisté à amplifier des deux manières décrites précédemment
deux PAC disponibles et déjà purifiés au laboratoire. Le marquage de ces produits
d’amplification et leur hybridation spécifique ou non sur des préparations chromosomiques
pourra déterminer l’efficacité de ces méthodes. Deux techniques de marquage des sondes
seront utilisées : par nick translation et par PCR.
• Les produits de DOP-PCR et du kit GenomiPhi seront marqués par nick translation,
• Le marquage par PCR consiste en l’incorporation de Biotine-dUTP directement au
cours de la PCR.
Le schéma 5 en annexe 4 présente un récapitulatif des différents marquages effectués.
11Matériel Et Méthodes
12I. LE KIT VYSIS « ToTelV
elVisionTM Multi-
Multi-probes Panel »
La technique de FISH fait partie du domaine de la cytogénétique moléculaire. Elle met
en jeu un ADN cible (chromosomes métaphasiques) et un ADN marqué, ou sonde
fluorescente, qui s’y hybride spécifiquement. Cette hybridation est réalisée sur des
préparations cellulaires fixées sur lames et se définit de ce fait comme hybridation in situ.
I.1. Obtention de métaphases
Les chromosomes sont obtenus à partir d’un prélèvement sanguin (600 µL) mis en
culture dans un milieu de croissance classique (7 mL) (RPMI 1640 Medium, GIBCOTM)
(additionné de 20% de sérum de veau) en présence d’un agent mitogène : la
phytothémaglutinine. Cette molécule a la propriété de stimuler les lymphocytes T quiescents
qui vont reprendre leurs divisions cellulaires. La culture sera bloquée au bout de 72 heures,
temps requis pour l’obtention d’un nombre optimale de mitoses, par un agent anti-mitotique,
la colchicine (Cf = 135 ng/mL). Du Bromure d’Ethidium (BET) (Cf = 1 g/L) est également
ajouté aux préparations, il contribue au bon étirement des chromosomes en s’intercalant dans
la double hélice de l’ADN. Le milieu est ensuite centrifugé (5 min, 1200 rpm), le culot est
repris dans une solution de KCl 0,075M, qui entraîne la lyse des cellules et le gonflement des
noyaux : c’est le choc hypotonique. La solution hétérogène obtenue est centrifugée (17 min,
1200 rpm, 37°C), et le culot, contenant les noyaux et les mitoses, est progressivement remis
en suspension et lavé dans une solution de fixateur (10 mL d’éthanol absolu et acide acétique
à 3 volumes pour 1) puis centrifugé (5 min, 1200 rpm). Cette étape de lavage est réalisée
plusieurs fois afin d’éliminer les débris cellulaires restants. Après la dernière centrifugation,
0,5 à 1 mL de fixateur sont conservés selon l’importance du culot, pour remettre les
préparations en suspension. 3 à 4 gouttes de cette suspension seront étalées sur lame de
microscope dans des conditions de température (21°C) et d’humidité (50%) permettant une
bonne dispersion des chromosomes.
Après étalement, les lames seront vieillies une nuit à température ambiante (ou 1 heure
à 60°C), afin d’améliorer la morphologie des chromosomes après contre coloration ultérieure
au DAPI.
I.2. Pré-Traitement à la pepsine
La pepsine est une protéase utilisée ici pour digérer les protéines restées fixées à l’ADN,
et enlever le voile cytoplasmique, visible au microscope à contraste de phase, entourant les
13chromosomes et gênant l’hybridation. Les lames seront incubées 10 minutes dans une solution
à 0,01 N d’HCl contenant 10 mg/L de Pepsine, à 37°C. Elles seront ensuite rincées 5 minutes
dans un bain de tampon PBS (NaCl 140mM, KCl 2,7mM, KH2PO4 1,5mM, Na2HPO4, 2H2O
6,5mM, pH 7.4), 5 minutes dans un bain de tampon PBS/MgCl2 5%, puis 2 minutes dans du
tampon PBS/MgCl2 5%/formaldéhyde1% et enfin 5 minutes dans du tampon PBS seul.
Les préparations sont ensuite déshydratées dans des bains d’éthanol de concentrations
croissantes (70%, 85% et 100%), 2 minutes à chaque fois. Les lames sont ainsi prêtes à être
hybridées.
I.3. Les sondes
Les sondes sont réparties en 15 mélanges :
I :1p vert, 1q rouge, XpYp rouge et vert, CEP X IX :9p vert, 9q rouge, 17q rouge et vert, CEP 17
aqua aqua
II :2p vert, 2q rouge, XqYq rouge et vert, CEP X X :10p vert, 10q rouge, 15q rouge et vert, LSI
aqua 15q22 aqua
III :3p vert, 3p rouge, 22q rouge et vert, LSI 22q11 XI :11p vert, 11q rouge, 18p rouge et vert, CEP 18
aqua aqua
IV :4p vert, 4p rouge, 21q rouge et vert, LSI 21q22 XII :12p vert, 12q rouge, 18q rouge et vert, CEP
aqua 18 aqua
V :5p vert, 5p rouge XIII :16p vert, 16q rouge
VI :6p vert, 6p rouge, 13q rouge et vert, LSI 13q14 XIV :19p vert, 19q rouge
aqua
VII : 7p vert, 7q rouge, 14q rouge et vert XV :20p vert, 20q rouge
VIII : 8p vert, 8q rouge, 17p rouge et vert, CEP 17
aqua
I.4. Hybridation des sondes
I.4.a. Hybridation
Les réactions se passent dans l’« HybriteTM (VYSIS)», spécialement conçu pour
l’hybridation in situ. C’est une chambre d’hybridation fermée et programmable, constituée par
une plaque chauffante sur laquelle sont déposées les préparations chromosomiques sur lames
(Document 6 en annexe 5).
5 lames préparées de la manière décrite ci-dessus sont observées au microscope à
contraste de phase afin de délimiter 3 zones sur chaque lame, riches en mitoses de qualité.
3µL de chaque mélange de sonde seront respectivement déposés sur chacune de ces zones et
recouverts par une lamelle circulaire (diamètre 1 cm) scellée par du rubber cément (schéma 7
en annexe 6).
14Une fois les lames ainsi introduites dans l’HybriteTM, la plaque chauffante élève la
température à 70 °C pendant 3 minutes pour dénaturer l’ADN et les sondes. La température
redescend ensuite doucement jusque 37°C, permettant aux sondes de s’hybrider
spécifiquement à l’ADN, pour s’y stabiliser 16 heures minimum.
I.4.b. Rinçage
Les lames sont incubées 2 minutes dans une solution de SSC (NaCl 3M, NaH2Ci
0,3M) 4× / NP-40 0,3% chauffée à 73°C±1°C (le NP-40 est un détergent qui va éluer les
sondes non hybridées ou faiblement liées à l’ADN et donc sur des régions non spécifiques),
puis 15 secondes dans une solution de SSC2× /NP-40 0,1% à température ambiante.
3µL du contre colorant DAPI (4’-6-diamidino-2-phénylindole) sont ensuite déposés
sur chaque zone et recouverts d’une lamelle 24×60mm pour en assurer la diffusion. Les lames
se conservent ainsi à 4°C et peuvent être congelées à -20°C.
I.4.c. Les fluorochromes
Quatre fluorochromes sont utilisés dans le kit :
Fluorochrome λmax d’excitation (nm) λmax d’émission (nm) Couleur
FITC (Isothiocyanate
490 520 vert
de fluorescéine)
Rhodamine 545 575 Rouge
Spectrum Aqua 433 480 Bleu
DAPI 355 450 Bleu
Remarque : la solution de DAPI contient un agent antifade qui limite les pertes de
fluorescence au cours du temps, les lames peuvent ainsi être conservées plus longtemps.
I.4.d. L’analyseur d’images
Les préparations chromosomiques hybridées sont donc excitées aux différentes
longueurs d’onde d’excitation de chaque fluorochrome, grâce aux filtres dont est équipé le
microscope à épifluorescence (Schéma 8 en annexe 7). La caméra numérique dont il est
équipé prend des clichés, ici 4, de chaque image restituée par chaque filtre. Le logiciel VYSIS
Quips® Lab Manager est un analyseur d’image composé de deux applications : Smart
Capture VP gère le traitement de l’image, File Maker Pro est une base de donnée regroupant
les dossiers des patients (document 9 en annexe 8).
15II.
II. CONCEPTION DE SONDES
SONDES FISH
II.1. Amplification des PACs
II.1.a. Les PACs utilisés
Le premier PAC utilisé est référencé RP13-377 G1, l’insert fait 146 633 pdb. Le
deuxième PAC utilisé est RP11 805 H4, l’insert fait 96 398 pdb. Ces deux PAC couvrent tous
les deux la région 11.22 du bras court du chromosome X (Xp11.22).
L’institut Sanger (http://www.sanger.ac.wk) propose notamment une banque de
données qui fournit une cartographie du génome humain sur chaque bande chromosomique,
les contigs, les différents marqueurs type STS (Séquence Tag Sites : ce sont des marqueurs
positionnels de l’ADN), les gènes et les différents vecteurs disponibles.
II.1.b. La DOP-PCR
Les amorces utilisées comportent en 3’ une séquence de 6 nucléotides spécifique de
séquences répétées de l’ADN humain. En amont de ces nucléotides figure une séquence
dégénérée de 6 autres nucléotides. La portion en 5’ de 10 nucléotides comprend un site de
restriction rare permettant un éventuel clonage ultérieur. [FIELGER et al., 2003].
DOP1 : 5’ CCGACTCGAGNNNNNNCTAGAA 3’
DOP2 : 5’ CCGACTCGAGNNNNNNTAGGAG 3’
DOP3 : 5’ CCGACTCGAGNNNNNNTTCTAG 3’
Les amorces sont synthétisées au service commun de biologie moléculaire du Centre
Hospitalier Universitaire de Nancy (CHU). La première PCR, est réalisée dans un volume
réactionnel de 50 µL contenant 5µL de tampon d’amplification concentré 10×, spécifique de
l’AmpliTaqGold polymérase (Applied Biosystem), 0,2 mM de dNTP (laboratoire Roche), 2
µM d’amorces DOP (1, 2, 3), 7,5% de DMSO, 4 mM de MgCl2, 3,75 unités d’AmpliTaqGold
polymérase, 50 ng de PAC et la quantité suffisante d’eau mQ pour 50µL. Les réactions de
polymérisation sont réalisées dans le thermo-cycleur BIORAD Icycler.
Après une dénaturation initiale à 94°C pendant 10 minutes, permettant l’activation de
l’AmpliTaqGold, les cycles d’amplifications sont : 10 premiers cycles comprenant une étape
de dénaturation à 94°C pendant 1min30s, une étape d’hybridation à 30°C pendant 2min30s,
une augmentation progressive à 0,1°C/sec jusque 72°C, et une étape d’élongation à 72°C
pendant 3min. Ces 10 cycles sont immédiatement suivis de 30 cycles de 94°C pendant 1min,
62°C pendant 1min30s, 72°C pendant 2min, et un dernier cycle à 72°C pendant 8min.
16Elle est suivie d’une deuxième PCR afin d’amplifier spécifiquement les produits de la
première PCR. En effet, l’amorce utilisée ici, appelée DOP4, est identique dans sa région 3’
des nucléotides de la région 5’ commune aux amorces DOP1, 2, et 3.
DOP4 : 5’ GGAAACAGCCCGACTCGAG 3’
La réaction se déroule dans le thermo-cycleur BIORAD Icycler. Elle est réalisée dans
un volume réactionnel de 60 µL contenant 6 µL de Tampon 10× spécifique à l’AmpliTaqGold
polymérase, 0,25 mM dNTPs, 2µM d’amorce DOP4, 3 U/µL d’AmpliTaqGold, 2,5mM de
MgCl2, 2 µL de produit de DOP-PCR. Après une dénaturation initiale à 95°C pendant 10min,
la réaction est composée de 35 cycles de 95°C pendant 1min, 60°C pendant 1min30s, 72°C
pendant 7min, suivis d’une étape finale de 72°C pendant 10min.
Les produits seront purifiés par le kit NucleoSpin®Extract de MACHEREY-NAGEL
avant d’être marqués par nick translation.
Remarque : dans le cas du marquage à la biotine par la deuxième PCR, celle-ci se
réalisera dans les mêmes conditions que sans biotine, mais avec l’AmpliTaq (Applied
Biosystem), 0,25 mM de dA/C/GTP, 0,125 mM de dTTP et 0,125 mM de biotine-16-dUTP
(laboratoires Roche) [JONVEAUX et al., 1996]. Les étapes de purification et de nick
translation ne seront alors pas nécessaires.
II.1.c. Utilisation du kit GenomiPhi
Ce kit utilise les propriétés d’amplification de l’ADN polymérase phi29. Il est
composé de quatre tubes : un mélange des hexamères aléatoires, un tampon réactionnel
contenant les dNTPs et les sels, conservés à –20°C, et un mélange d’enzymes conservé à
–70°C. Les manipulations se font dans la glace entre 0°C et 4°C. 1µL de matrice d’ADN
(PAC purifié à 1ng/µL minimum) sera ajouté à 9µL de la solution contenant les amorces
(hexamères). Ce mélange est dénaturé à 95°C pendant 3 minutes, refroidi dans la glace et
ajouté à 9µL du tampon de réaction et 1 µL d’enzyme. Ce mélange est incubé 16 à 18 heures
à 30°C.
A la fin de ces 18 heures, l’action de l’ADN polymérase phi 29 est stoppée par la
chaleur en incubant le mélange réactionnel 10 minutes à 65°C. Ce mélange peut maintenant
être purifié (NucleoSpin®Extract de MACHEREY-NAGEL) puis dosé en quantité d’ADN
pour être marqué et hybridé.
17II.2. Marquage des sondes
Dans le cas du marquage par PCR, cette étape n’est pas réalisée.
Les produits de DOP-PCR, de deuxième PCR et du kit GenomiPhi (purifiés) sont
marqués par Nick Translation avec le kit BIONICK Labelling System (Invitrogen) (Schéma
10 en annexe 9)
La concentration d’ADN avant marquage sera dosée par fluorescence grâce au réactif
de Hoescht 33258 qui est un intercalant de l’ADN et qui n’est fluorescent qu’une fois inséré
dans la double hélice. Le signal de fluorescence obtenu est directement proportionnel à la
quantité d’ADN présent. Il suffit d’étalonner l’appareil avec une solution d’ADN de
concentration connue.
II.3. Précipitation de l’ADN marqué.
A ce stade, les sondes sont marquées, soit directement par PCR, soit indirectement par
nick-translation. Mélanger 1 µg d’ADN marqué avec 45µg d’ADN Cot-1 (généralement 40 à
45 fois la quantité d’ADN marqué), ajouter 1/10 volume d’acétate de sodium 3M, pH5,2, et
2,5 volumes d’éthanol absolu à –20°C. Ce mélange est incubé à –20°C pendant 10 minutes
avant d’être centrifugé 30 minutes à 14 500 rpm à +4°C.
Le surnageant est éliminé par retournement, et le culot lavé avec 500µL d’éthanol 70%
à –20°C par centrifugation 5 minutes à 14 500 rpm. Ce culot est ensuite mis à sécher à
l’évaporateur sous vide, 1 minute maximum pour ne pas endommager les sondes. Sec, il est
resuspendu dans 20 µL de tampon d’hybridation (0,1g/mL de sulfate de dextran, 50%
formamide, SSC2×, 1% Tween20, H2O distillée). On ajoutera également une sonde
commerciale témoin XqYq (1µL).
III. HYBRIDATION DES SONDES SUR LAME
L’étalement et le prétraitement des lames est réalisé de la même manière que pour
l’hybridation avec le kit ToTelVisionTM.
III.1. hybridation
Les lames sont incubées 2 minutes dans une solution de formamide70%/SSC2×, pH7-
7,5 à 72±1°C, puis immédiatement et successivement 2 minutes dans trois bains d’éthanol
70%, 85% et 100% à –20°C. Elles sont ensuite séchées à l’air libre. Les sondes sont
dénaturées 5 minutes 73°C puis incubées 2 heures à 37°C.
1810 à 13µL de mélange de sondes dénaturées sont déposés sur une région de lame
dense en mitoses, une lamelle 24×32mm y est déposée et scellée à l’aide de rubber cément.
Les lames seront ensuite mises en chambre humide à 37°C de 18 à 24 heures.
III.2. Rinçage et détection immunochimique
Les lames sont lavées 2 minutes dans une solution de SSC4×/0,3% NP-40 à 72±1°C,
15 secondes dans une solution de SSC2×/0,1% NP-40 à température ambiante, puis égouttées.
150µL de BN block [8,4 g/L de bicarbonate de sodium, 0,25% NP-40, 5% poudre de lait, H2O
distillée] y sont déposés et recouverts par une lamelle 24×60mm souple. Après 10 minutes
d’incubation à température ambiante, les lames sont encore égouttées.
60 µL d’avidine marquée à la fluorescéine (FITC) y sont alors déposés avant de les
incuber 10 minutes à 37°C en chambre humide. Elles sont ensuite lavées trois fois 2 minutes
dans une solution BN [8,4 g/L de bicarbonate de sodium, 0,25% NP-40, H2O distillée]. Cette
opération (marquage + lavage) est répétée deux autres fois, mais respectivement avec des
anticorps biotinylés anti-avidine, et de l’avidine marquée FITC (Schéma 11 en annexe 10) A
la fin du dernier lavage, les lames sont mises à sécher à l’air libre et à l’abri de la lumière.
9µL de DAPI sont ensuite déposés sur la lame que l’on recouvrira d’une lamelle rigide
24×60mm. Les lames seront ainsi concervées à +4°C.
Un récapitulatif des étapes majeures est présenté en annexe 11.
19RESULTATS
20I. LE KIT ToTelVision
ToTelVisionTM Multi-
Multi-probes Panel
Ce kit est composé de 15 mélanges de sondes télomèriques, centromèriques et locus
spécifique. Les sondes centromèriques (CEP) et locus spécifiques (LSI) servent de témoin de
reconnaissance des chromosomes. En effet, le contre colorant DAPI s’intercale dans le petit
sillon des régions riches en A et T de l’ADN. Il s’intercale également avec une plus faible
affinité dans le grand sillon de la double hélice des régions riches en G et C. On obtient ainsi
une ébauche de marquage en bandes G des chromosomes permettant leur identification.
Cependant, l’utilisation des sondes témoins est plus rigoureuse.
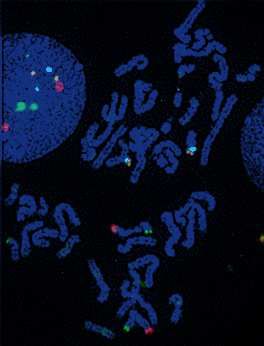
Sur 15 dossiers traités durant ce stage, je n’ai mis en évidence aucune anomalie des
régions subtélomèriques. Il est à noter que ce genre d’anomalie est retrouvé dans 4 à 7% des
cas de retards mentaux idiopathiques. Les images d’hybridation in situ correspondant à
l’analyse chromosomique d’un patient sont exposées en annexe 13.
Ces résultats ont été traités par l’application SmartCaptureVP du logiciel
®
Quips LabManager qui permet d’amplifier les signaux d’hybridation et d’atténuer le bruit de
fond éventuel, résultant d’un rinçage non stringent des lames.
II. Les sondes conçues par DOP-
DOP-PCR et par le kit Genomiphi
II.1.Amplification d’ADN par DOP-PCR
II.1.a. Les produits de la première PCR
L’utilisation de l’ampliTaq type Gold (Applied Biosystem) a été privilégiée ici pour sa
processivité, permettant d’amplifier des fragments avec le minimum d’erreur, de manière à
obtenir des sondes de la meilleure qualité possible. De plus, le tampon d’amplification
spécifique de l’ampliTaqGold ne contient pas de MgCl2, il est fourni à part, ce qui permet
d’en ajuster la concentration. J’ai donc testé la DOP-PCR en utilisant différentes
concentrations de MgCl2 et d’enzyme (Document 13 en annexe 12), ce qui a permis de définir
les conditions optimales d’amplification : 3,75 unités d’enzyme dans 50 µL (0,075 U/µL) et 4
mM de MgCl2, conditions qui sont un bon compromis entre l’intensité des bandes sur-
amplifiées et celle de la traînée, cette intensité étant proportionnelle à la quantité d’ADN
amplifiée.
21Les produits d’amplification par les amorces 1, 2 et 3 des deux PACs font apparaître
sur gel d’agarose une traînée importante, compris entre 0,2 kb et 2,6 kb environ, avec des
bandes bien marquées de tailles variables selon l’amorce utilisée. Les documents 14 en
annexe 14 et 15 en annexe 15 nous montrent notamment que l’amorce DOP2 donne lieu à la
sur-amplification des plus grands fragments amplifiés par DOP-PCR, et DOP3 de fragments
plus petits.
On obtient un profil de migration reproductible propre à chaque amorce DOP ou
mélange d’amorces, pour chaque PAC et dans les mêmes conditions de PCR.
On voit que les blancs présentent une traînée qui pourrait faire penser à une
contamination. La même expérience a été réalisée en changeant tous les substrats de la PCR :
amorces, tampon d’amplification, MgCl2, dNTP, enzyme : les blancs présentaient toujours
une traînée. On peut par contre émettre une hypothèse : les amorces, étant en partie
dégénérées, et n’ayant aucune matrice d’ADN, se polymériseraient et formeraient un réseau
qui s’amplifierait aléatoirement. L’utilisation de DMSO, agent minimisant les structures
secondaires et optimisant les PCR [PROMEGA NOTES, 1998], améliore d’une part le
rendement de la DOP-PCR, mais contribue également à une diminution notoire de l’intensité
de la traînée dans les blancs (Document 16 en annexe 16).
De plus, le profile de migration du PAC dans un gel d’agarose 8% ne montre pas la
présence d’ADN contaminant. L’amplification du PAC par les amorces du gène MECP2,
localisé en Xq28 (Document 17 en annexe 17), réfute l’hypothèse d’une contamination par de
l’ADN génomique. Ceci est confirmé par une hybridation in situ des produits de DOP-PCR
spécifique.
II.1.b. Les produits de deuxième PCR
Les amorces DOP4 permettent théoriquement l’amplification spécifique des produits
de la DOP-PCR, la traînée réapparaît donc entre 0,2 et plus de 2,6 kb, mais tous les fragments
amplifiés par DOP-PCR ne sont pas ré-amplifiés. Les profils de migration obtenus sont
également reproductibles pour les mêmes conditions de PCR.(Doc 18 en annexe 17). Dans le
cas du marquage à la biotine par PCR, on peut observer un retard de migration de l’ensemble
des fragments amplifiés, sans modification du profil de migration (Doc 19 en annexe 18).
22II.2. Amplification d’ADN par le kit Genomiphi
Ici aussi l’amplification est aléatoire, on observe donc logiquement une traînée mais
sans bandes. Celui-ci est de taille comprise entre 2 et 20 kb (Doc 20 en annexe 18). L’ADN
polymérase Phi29 permet donc l’amplification de fragments de grandes tailles.
Ces différents produits sont purifiés afin d’éliminer les nucléotides avant nick
translation, et des nucléotides marqués non incorporés par PCR dans le cas du marquage par
PCR.
II.3. Marquage des produits d’amplification
Les fragments marqués de DOP-PCR et de deuxième PCR sont de taille minimum (0,2
kb environ), ceux du kit Genomiphi entre 0,4 et 1 kb (Doc 21 en annexe 19) Ces sondes sont
précipitées, lavées puis reprises dans du tampon d’hybridation.
II.4. Hybridation
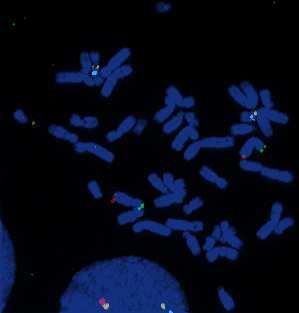
Les sondes témoins XpYp rouges sont spécifiques respectivement des régions
subtélomèriques des chromosomes X et Y. Les sondes marquées par nick translation (en vert)
des produits de première PCR et deuxième PCR s’hybrident spécifiquement à la région
Xp11.22 avec un bon signal de fluorescence (Documents 22 en annexe 20 et 23 en annexe
21). Par contre, les produits marqués d’amplification du kit Genomiphi font apparaître des
signaux non spécifiques, sur des chromosomes différents (Document 24 en annexe 22).
23DISCUSSION
24I. Le kit ToTelVisionTM multi-
multi-probes panel
I.1. Les sondes
Leur composition n’est pas divulguée, mais pour une bonne hybridation, les mélanges
de sondes doivent contenir plusieurs éléments essentiels :
de la formamide qui contribuera à diminuer la température de dénaturation Tm
de l’ADN et donc à abaisser la température de la réaction d’hybridation,
du Sulfate de Dextran, qui favorise l’association entre molécules complètement
ou partiellement hybridées pour former un maillage qui augmente le taux d’hybrides formés et
donc le signal au site d'hybridation,
Du SDS (Sodium-Dodécyl-Sulfate), détergent qui limite les hybridations
aspécifiques,
Une concentration en sels de sodium particulière. En effet, les ions
monovalents comme le sodium Na+ neutralisent les charges négatives (répulsives) des
groupements phosphates de l’ADN et de la sonde, et donc favorisent l’hybridation.
Remarque1 : l’équation de Thomas et Dancis (1993) permet de caractériser la température de
fusion Tm et donc la stabilité des hybrides :
Tm = 81.5 + 16.6 (log M) + 0.41 (% GC) – 820/L –0.6 (% F)
Où M est la force ionique (moles/litre de sels), % GC est le pourcentage en mole de
nucléotides G et C dans la sonde, L la longueur de la sonde en bases, et % F le pourcentage
de formamide dans la solution. D’après cette équation, une force ionique élevée stabilise les
hybrides (tout comme un pourcentage élevé en bases G et C, et une longueur importante de
la sonde). Remarque2 : Cette équation peut se simplifier pour des oligonucléotides de 11 à
20 pb par la relation :
Tm = 4 (G + C) + 2 (A + T)
De l’ADN humain appelé Cot-1 spécifique des séquences répétées du génome
humain. Il va donc s’y hybrider et ainsi empêcher une hybridation aspécifique qui pourrait
masquer le signal spécifique de la sonde.
I.2. les étapes de rinçage
Elles sont très délicates et demandent une certaine maîtrise des techniques
d’hybridation in situ. Les signaux de fluorescence doivent être très spécifiques des régions
25d’intérêts, ainsi les bains de rinçage éliminent les sondes non hybridées ou hybridées de façon
aspécifique. Ces dernières peuvent être de même intensité que le signal spécifique à cause
d’un rinçage non stringeant (trop rapide ou à mauvaise température), rendant le diagnostique
difficile, voir incertain. Inversement, un rinçage trop long atténue l’intensité du signal de
fluorescence jusqu’à l’extinction.
II. Les sondes conçues par DOP-
DOP-PCR
Les amorces de la DOP-PCR utilisées ici permettent une amplification générale de
l’ADN humain, ceci sans spécificité de séquence, et préférentiellement par rapport à l’ADN
bactérien du vecteur. En effet, ces amorces ont en 3’ une séquence hexamèrique retrouvée
avec une moindre fréquence dans l’ADN d’E.coli (f0,4 soit 1 événement tous les 2,5 kb) [FIELGER
et al., 2003]. Comme ces séquences sont dispersées dans le génome humain, l’amplification
commence simultanément à plusieurs endroits. Les résultats obtenus ont mis en évidence
l’efficacité supérieure de la DOP-PCR par rapport au kit Génomiphi dans la conception de
sondes locus spécifique.
II.1. Produits de la DOP-PCR
La séquence des amorces sont en partie dégénérées, cependant l’hybridation est
permise à 30°C durant les dix premiers cycles de la DOP-PCR et par l’augmentation très
progressive de la température (0,1°C/sec) avant la phase d’élongation. Ceci a pour but de
stabiliser les duplex ADN-amorces dégénérées et permettre à la polymérase de commencer
l’élongation avant que ces hybrides ne soient trop déstabilisés par la température. Les 30
cycles suivants servent à amplifier les produits des 10 premiers cycles : en effet, les fragments
générés au cours de ces 10 cycles comportent à leurs extrémités les amorces et leur séquence
complémentaire. La température des cycles suivants est augmentée (62°C) de façon à ne
permettre uniquement que l’hybridation des amorces entières.
II.2. La deuxième PCR
Les produits d’amplification par l’amorce DOP 2 présentant des fragments de tailles
variées, ce sont ces derniers qui ont été marqués à la biotine en deuxième PCR. A titre
comparatif, le marquage par nick translation a été réalisé sur les mêmes produits
d’amplification par l’amorce DOP2. Mais on a pu également constater que l’amorce DOP3
26Vous pouvez aussi lire

















































