ONCO-HÉMATOLOGIE PÉDIATRIQUE - LES PATHOLOGIES LES PLUS FRÉQUENTES Véronique LAITHIER - Oncolie
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
ONCO-HÉMATOLOGIE PÉDIATRIQUE
LES PATHOLOGIES LES PLUS FRÉQUENTES
Véronique LAITHIER
EPIDEMIOLOGIE 1 % des cancers Incidence: 132 cas/an/million d’enfants 1700 nouveaux cas < 15 ans en France 50 % < 5 ans 2000/ an < 18 ans Répartition tumeurs solides variable selon âge 2éme cause décès < 15 ans après AVP
EPIDÉMIOLOGIE
EPIDÉMIOLOGIE: LEUCÉMIES
1er cancer de l’enfant (30%)
LAL: Leucémies Aigües Lymphoblastiques: 80 % âge: pic 2-5 ans
LAM: Leucémies Aigües Myéloblastiques: 17-18 % tous âges
Leucémies chroniques: 2- 3 %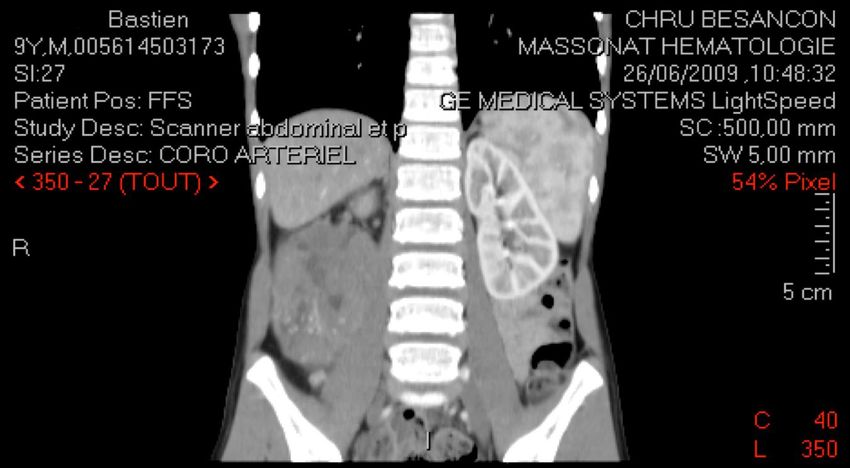
EPIDEMIOLOGIE:
TUMEURS CÉRÉBRALES (SNC)
25 % des Kcs
1ère cause de tumeur solide en pédiatrie et en raison des progrès tt pour
les LA, la 1ère cause de mortalité par Kc
Grande hétérogénéité et posent des pbs différents selon
l ’âge de survenue
leur caractère topographique
leur type histologique et grade de malignité
Age de survenue: nouveau né à adolescence
facteur de la présentation clinique
facteur important des effets délétères à long terme
Objectif thérapeutique: amélioration du pronostic vital avec diminution
de la morbidité neurologique, intellectuelle et endocrinienne: qualité de
vie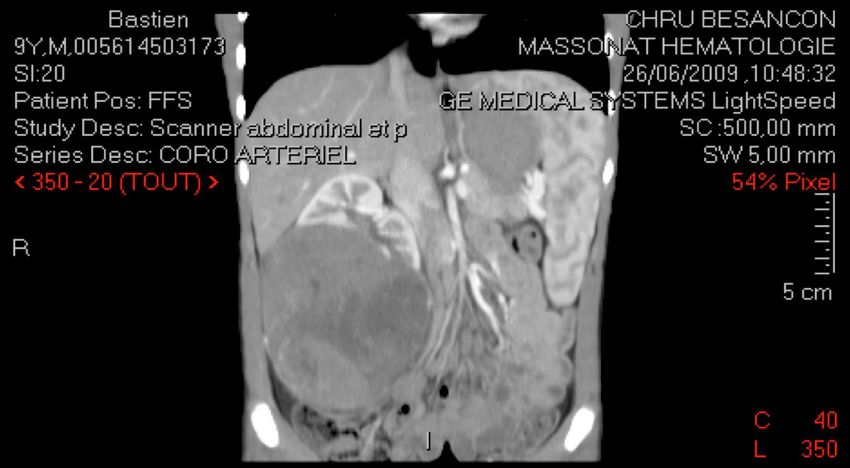
EPIDÉMIOLOGIE: TUMEURS SOLIDES SELON
L’AGE (HORS SNC)
< 1 an : néphroblastome, rétinoblastome, hépatoblastome…
1-4 ans : neuroblastome, néphroblastome, rhabdomyosarcome,
hépatoblastome
5-9 ans : lymphome, rhabdomyosarcome…
10-14 ans : lymphome, tumeur osseuse (Sarcome d’Ewing,
ostéosarcome) , sarcome tissu mou, …
AJA: lymphome, tumeurs osseuses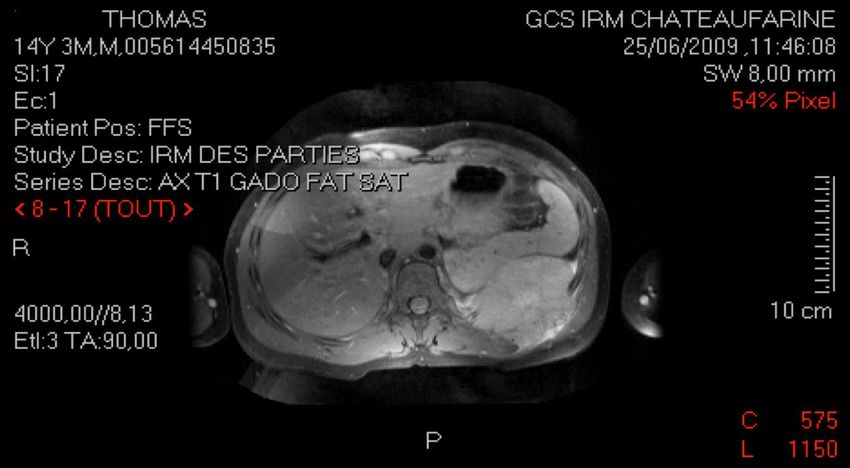
DIFFERENCES ADULTE/ENFANT Enfant: Organisme en Croissance 2 interlocuteurs: enfant + 2 parents
DIFFÉRENCES CANCER ADULTE/ENFANT
Localisation :
Sein, poumon, peau, tube digestif : rare en pédiatrie
Rein, SNC, os, tissus mous, système nerveux
sympathique, œil, organes génitaux, foie…
Histologie :
Carcinome : quasi-inexistant
2/3 tumeurs solides = tumeurs embryonnaires
(« blastome »). Rôle de gènes impliqués dans la
croissance du fœtus.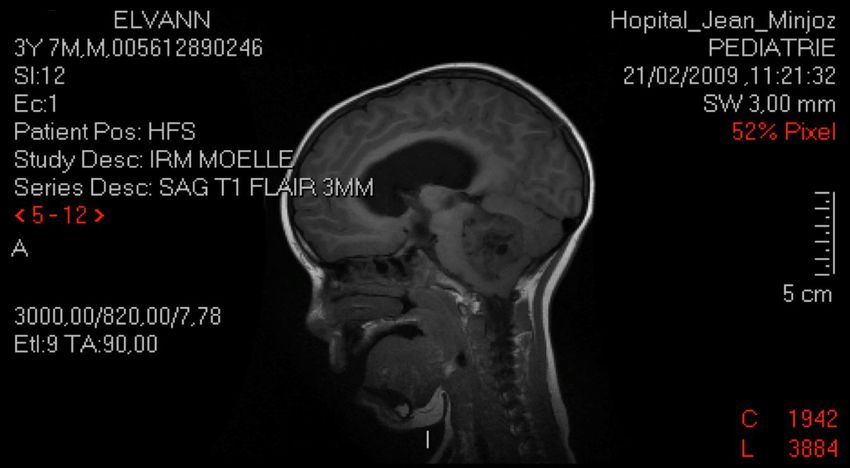
DIFFÉRENCES CANCER ADULTE/ENFANT
Facteurs de risque environnementaux (tabac, amiante) :
non-impliqués en pédiatrie
Reconnus (< 5 % des cancers pédiatriques)
Radiations ionisantes fortes doses
Chimiothérapie
Syndromes génétiques de prédisposition (Li Fraumeni, Fanconi,
Rétinoblastome, Wiedemann-Beckwith, Ataxie-Telangiectasie,
NF1…)
Agents infectieux : EBV +++
Suspectés :
radiations faible dose, lignes HT
Pollution, pesticides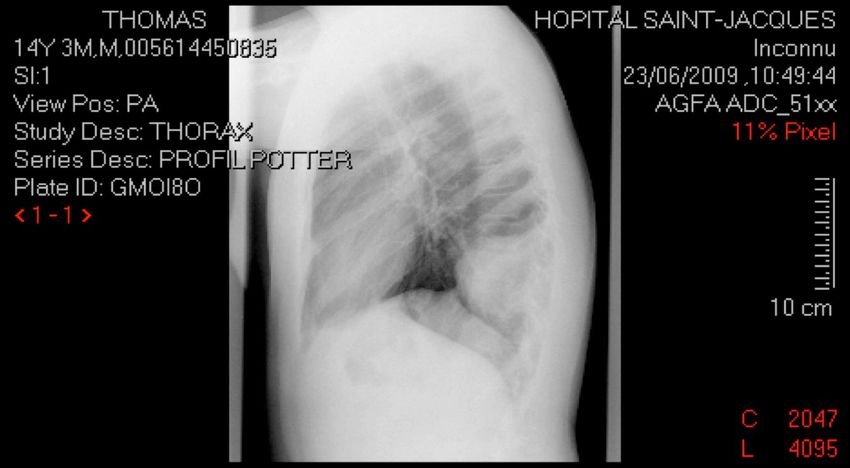
DIFFÉRENCES CANCER ADULTE/ENFANT Présentation clinique : État général souvent conservé en pédiatrie Croissance tumorale rapide (tumeurs embryonnaires) Extension peut être métastatique d ’emblée Pronostic : 75 à 80 % de guérison Chimiosensibilité/ radiosensibilité Meilleure tolérance chimiothérapie Problématique du long terme en pédiatrie Qualité de vie: séquelles neurocognitives Cancer secondaire, fertilité, séquelles: cœur
TAUX DE GUERISON: 75 % GRÂCE A LA COOPERATION
MULTICENTRIQUE ET AUX PROGRES THERAPEUTIQUES
-Très bons cas = 35% Optique= désescalade tt
LAL: 90 %, Nephro: 90%, LMH: 92%, Burkitt: 90%, NB localisé:
80%
-Cas + difficiles = 30% améliorer la survie et diminuer les
séquelles
Ostéosarcome: 70%, Ewing:60%, tu mésen: 60%
-Pbs non résolus: 35%
NB méta: 50%, TC: 50% séquelles neuropsyDIAGNOSTIC
Retard diagnostique fréquent :
Symptômes peuvent avoir allure banale
Etat général est longtemps conservé
Symptômes persistants
Rares cas urgence médicale :
Détresse respiratoire (compression médiastinale, leucostase
pulmonaire)
Compression médullaire, torticolis
Convulsion
Hémorragies…
Dispositif d’annonce diagnostiqueBILAN CANCER ENFANT
Bilan local de la tumeur :
Imagerie
Biopsie le + souvent
Technique la moins invasive possible
Examen histologique
Biologie moléculaire
Tumorothéque
Bilan d’extension
Bilan pré-thérapeutique (cœur, foie, rein…)
Préservation fertilité si indiquéeTRAITEMENT DES CANCERS DE L’ENFANT: RCP
INTER-RÉGIONALE (OIR) INCA
Objectifs :
Proposer la même qualité de prise en charge pour tous les patients
Faire profiter le patient des progrès médicaux : moyens
diagnostiques, nouvelles drogues, radiothérapie innovante
Définir la stratégie de traitement de façon optimaleTRAITEMENT DES CANCERS DE L’ENFANT
Collaboration nationale (SFCE) et internationale (SIOP et
Mondiale)
Médecins, chirurgiens, biologistes, pharmaciens,
radiothérapeutes, radiologues, anatomo-pathologistes,
psychologues
Regroupés en comités
Protocoles nationaux ou internationaux
Essais thérapeutiques
> 2/3 enfants inclus, consentement éclairé
Harmonisation des traitements
Progrès prise en charge
Meilleure compréhension
Nouvelles molécules
Désescalade thérapeutique pour tumeurs de bon pronosticTRAITEMENT DES CANCERS DE L’ENFANT
Chirurgie :
Meilleur traitement des tumeurs localisées
Parfois chirurgie de métastases
De + en + après chimiothérapie néoadjuvante :
Évaluation chimiosensibilité
Intervention moins compliquée :
limite le risque pour organes du voisinage
diminue risque de rupture tumorale per-opératoire
diminue indications d’amputation
Discussion en RCT nationale pour tu osseusesTRAITEMENT DES CANCERS DE L’ENFANT
Chimiothérapie :
Souvent utilisée en 1er :
Soit exclusivement (leucémies, lymphomes)
Soit en néoadjuvant
Soit en adjuvant
Intensification possible (effet-dose) + autogreffe de CSH
• Séquelles : cœur, croissance, cognition, audition, 2ème tumeur,
fertilité (risque non-majoré pour descendance)TRAITEMENT DES CANCERS DE L’ENFANT
Radiothérapie
Utilisée pour contrôle local de tumeurs si chirurgie impossible ou
en complément pour tu agressives, métastases
TC : ATTENTION < 5 ans car séquelles neurocognitives, hormonales
Indications
Séquelles à long terme ++ (croissance, tumeur secondaire,
insuffisance thyroidienne…)
Amélioration des techniques (protons, tomothérapie, stéréotaxie)
pour limiter effets irradiation tissus sains
Discussion des dossiers en RCT nationale et contrôle qualitéTRAITEMENT DES CANCERS DE L’ENFANT
Molécules ciblées
Inhibiteur de tyrosine kinase dans LAL Phi +, LMC
Inhibiteur de ALK dans ALCL, NB
Inhibiteur de mTOR dans NF plexiformes
BIOMEDE dans DIPG
MAPPY ACTS
Séquelles à long terme ?HEMATOLOGIE-ONCOLOGIE PÉDIATRIQUE Groupe hétérogène de tumeurs différentes de celles de l’adulte Chaque tumeur est une maladie rare Meilleure connaissance = diagnostic précoce Amélioration taux guérison grâce à collaboration nationale et internationale Amélioration soins de support = objectif primordial Amélioration qualité de vie à long terme
AVENIR APRÈS LA MALADIE ET LE TRAITEMENT
Surveillance prolongée
Rechute
Séquelles
Fertilité, aide à la procréation
Insertion sociale/professionnelle
Second cancerCAS CLINIQUE 1
Donovan, 4 ans :
Fièvre 39°C depuis 5j, pâleur, asthénie
Boîterie depuis 10 j
Examen clinique :
pétéchies tronc, membres inférieurs et palais, pâleur cutanée, asthénie,
tachycardie à 125/min avec SS
HSM, ADP cervicales + inguinale
OMA D, herpès labial
NFS : 1 900 leucocytes dont 13 % de lymphoblastes, Hb 4,1 g/dL et
19 000 plqt
Myélogramme : 95 % de lymphoblastes : Leucémie aiguë
lymphoblastique BLAL
1er cancer de l’enfant (30 %)
Blocage hématopoïèse par une prolifération monoclonale de
cellules : blastes
Lymphoblastes : LAL > 80 % (pic 2-5 ans)
LAM, leucémies chroniques : 20 % (tous âges)LAL
Facteurs pronostiques :
Sexe masculin
< 1 an ou > 10 ans
Hyperleucocytose > 50 G/L
Phénotype T
Anomalies cytogénétiques, biologie moléculaire
Réponse au traitement: Cortico-résistance, maladie résiduelle
Atteinte méningée, gonades, médiastin
Sd tumoral importantLAL Clinique : AEG, fièvre, sueurs nocturnes Sd d’insuffisance médullaire : Anémie : pâleur, asthénie, dyspnée Thrombopénie : purpura, hémorragie… Neutropénie : infections… Sd tumoral : ADP, HSM Douleurs osseuses Hypertrophie gingivale
LAL
Traitement « lourd » durant 5 à 12 mois
Traitement entretien pour un total de 2 ans
Pronostic :
LAL : > 80 % survie sans évènement à 5 ans
LAM : 50-70 %CAS N°2
Elvann: 5 ans
HTIC: Céphalées + vomissements en jet matinaux,
amaigrissement
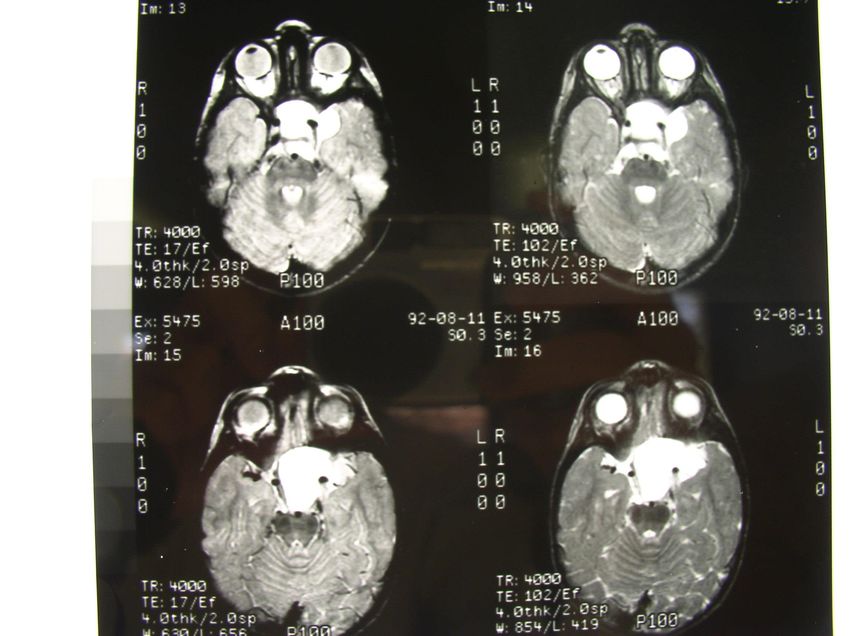
Troubles équilibre, strabisme acquisMÉDULLOBLASTOME
MÉDULLOBLASTOME
MÉDULLOBLASTOME: 10-20 % TC
PNET FP: tumeur maligne ++ (blastème) avec risque de dissémination
dans le SNC par le LCR
TT et pronostic diffèrent selon l’âge, résidu, type histologique, atteinte
LCR, métastases et biologie moléculaire: 4 ss types
Tu chimio et radio sensible
Traitement: chir, RT CS +/- CT grands enfants, CT petits (CT conv +/- HD
pour éviter ou limiter RT)
En 70, taux de guérison = 30 à 40%, actuellement 60-80%: 75-80%
pour RS et < 50% pour HR
Protocole national d’étude de l’évaluation neuropsy: difficultés scolaires
croissantes: lenteur, troubles de la mémoire, difficultés de concentrationCAS N°3 Anne 9 mois Cassure pondérale Nystagmus
GLIOME DES VOIES OPTIQUES
5 % des TC de l’enfant
65 % < 5 ans
Astrocytome de bas grade, mais comportement non prévisible: de
régression spontanée à atteinte visuelle et/ou neurologique majeure,
endocrinienne pouvant conduire au décès
30 % NF1
SC: HTIC (30%), déficience visuelle (50%), cachexie diencéphalique (14%)
Traitement: chimiothérapie !!!: plusieurs lignes parfois pour éviter ou
retarder la RT
PFS: 40 %, OS: 90 %, RFS: 61 %P.Keihues et al : WHO classification of brain tumours
.Table 2 World Health Organization (WHO) grading system (malignancy scale) of CNS tumours.
CLASSIFICATION DES TC
Tumour Group
Astrocytic tumours
Tumeur Type
Subependymal giant cell
Grade I
Grade II Grade II Grade I
Pilocytic
Low grade
Pleomorphic xanthoastrocytoma
Anaplastic
Glioblastoma
Oligodendrogliomas Low grade
Anaplastic
Oligo - astrocytomas Low grade
Anaplastic
Ependymal tumours Subependymoma
Myxopapillary
Low grade
Anaplastic
Choroid plexus tumours Papilloma
Carcinoma
Neuronal/glial tumours Ganpliocytoma
Ganglioglioma
Desmoplastic infantile ganglioglioma
Dysembryoplastic neuroepithelial tumour
Central neurocytoma
Pineal tumours Pineocytoma
Pineocytoma / pineoblastoma
Pineoblastoma
Embryonal tumours Medulloblastoma
Other PNETs
Medulloepithelioma
Neuroblastoma
Ependymoblastoma
Cranial & spinal nerve tumours Schwannoma
Malignant peripheral nerve sheath tumour
Meningeal tumours Meningioma
Atypical meningioma
Papillary meningioma
Hemangiopericytoma
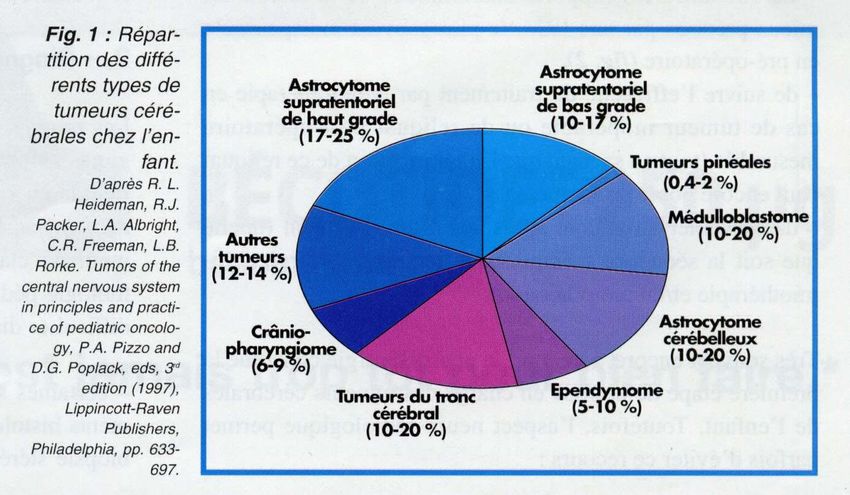
Anaplastic meningioma RÉPARTITION DES TC DE L ’ENFANT
LOCALISATION ET RÉPARTITION: TC
Développement dans la totalité du SNC (90% cerveau, 10 % moelle épinière)
mais 55% sont dans la FP (=1/10 du volume sustent)
Siège en fonction de l ’âge:
nourrisson: sus-tentoriel
enfant: infra-tentoriel
adolescent: sus-tentoriel
Histologie en fonction de l ’âge sauf astrocytomes à tout âge
0 1 3 5 10 15 ans adulte
CPC épendymome glioblastome
PNET médulloblastomePRÉSENTATION CLINIQUE: RETARD++
Dépend de la localisation tu et de l ’âge de l ’enfant
HTIC: 1ers signes tu FP, hydrocéphalie
Signes de focalisation: 1ers signes tu sustent
Ù FP:
Ù cervelet: ataxie, troubles coordination, nystagmus, paralysie nerfs crâniens
surtout VI, torticolis (engagement),
Ù TC: déficit moteur des voies longues, troubles déglution
Ù Tu hémisphérique: HTIC, déficit moteur, convulsion, svt silencieuse
longtemps (zones silencieuse du cerveau), trbles comportement
(frontale)
Ù Chiasma, hypothalamus: AV, CV, altérations fonctions
hypophysaires (taille, poids, DI, puberté)
Nourrisson: PC, regard en coucher de soleil, retard de la marche,
VISION, cachexie diencéphaliqueTUMEURS CÉRÉBRALES:
Challenge diagnostique, RCP +++
Challenge thérapeutique: chir, CT, RT, mol ciblées
Grande diversité de pronostic: survie globale: 73% à 5 ans
Astrocytome pilocytique: 87%
Médulloblastome: survie à 5 ans: 60 à 70%
Ependymome: survie à 5 ans: 40 à 50%
PNET sus tent: 34%
TGM: 92%
Gliome infiltrant du TC: médiane de survie de 9 mois à 2 ans
ATRT: 35%
Neuro-oncologie pédiatrique en cours de développement:
Diagnostic: neuroradiologie, biologie moléculaire
Pronostic: biologie moléculaire
Thérapeutique: nouvelles modalités thérapeutiques et essais tt
Evaluation neuropsychologique et qualité de vieDIAGNOSTIC : TUMEUR ABDOMINALE
Circonstance découverte :
Parents lors toilette « gros ventre dur »
Examen médical systématique
Douleurs abdominales
Souvent asymptomatique
Etiologies :
Neuroblastome, néphroblastome
Lymphome
Hépatoblastome, tumeur germinale maligneDIAGNOSTIC : TUMEUR ABDOMINALE
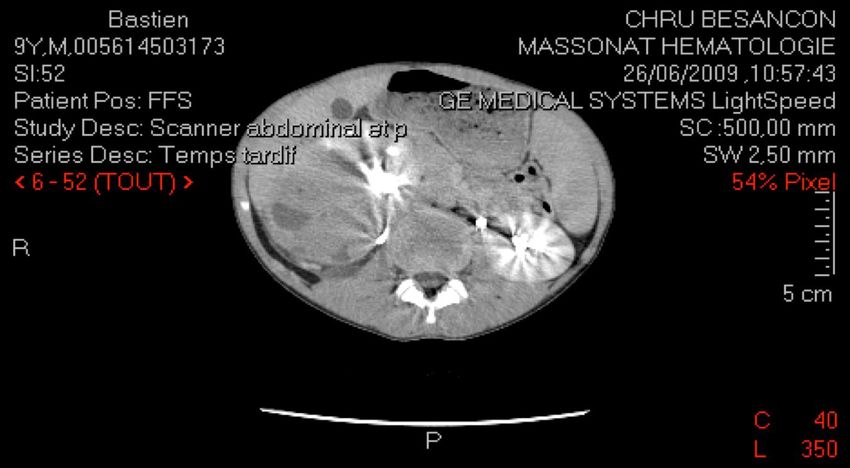
CAS N°4
Bastien, 9 ans
Douleur brutale flanc droit + fièvre à 39°C -> Sd appendiculaire
Interrogatoire :
douleur hanche droite depuis 1 mois, asthénie depuis 15 j
examen clinique :
Palpation masse volumineuse dans le flanc droit (10 cm)
état général conservé.CAS N°4
CAS N°4
CAS N°4
NÉPHROBLASTOME (ou tumeur de Wilms) tumeur maligne embryonnaire au sein parenchyme rénal tumeur du rein la plus fréquente chez l'enfant (> 90%) 5% de l'ensemble des cancers de l'enfant (1/10 000 naissances) entre 1 et 5 ans (98 % avant 7 ans) 10% association sd polymalformatifs
NÉPHROBLASTOME
Masse abdominale : tumeur volumineuse, antérieure,
ferme, lisse, peu mobile, indolore, augmente
rapidement de volume, fragile +++
Hématurie (25 %)
HTA (30 à 60%) liée à une sécrétion de rénine par la
tumeur ou compression des vaisseaux rénaux
Fièvre
Douleurs abdominales (distension, hémorragie intra-
tumorale)
malformations associées : hémi-hypertrophie
corporelle, l’aniridie, Sd WAGR, Sd Wiedmann-BeckwithNÉPHROBLASTOME
Traitement :
Chimiothérapie pré-opératoire : 1 mois
Urétero-Néphrectomie élargie (partielle dans de rares indications)
Radio et/ou Chimiothérapie post op selon stade
90 % survie globale (100% pour stades favorables 63 %, pour
haut risque)CAS N°5 Yann 11 ans Douleur pariétale thoracique G depuis 5 j, vomissements et fébricule depuis 2 j Examen clinique normal RP : masse thoracique prévertébrale G TDM C-TAP : masse hétérogène de 10cm au niveau de la gouttière vertébro-costale gauche de D5 à D11 avec extension au niveau du trou de conjugaison D7-D8 et érosion osseuse au contact
CAS N°5
CAS N°5
NEUROBLASTOMES
= tumeur maligne dérivée des cellules originaires des crêtes
neurales ; rachis, médullosurrénale, périartériel
10% des tumeurs solides de l’enfantSYMPTÔMES LIÉS AU PRIMITIF
tumeurs abdominales : Masse abdominale, douleur
abdominale chronique, brutale si saignement intra ou péri-
tumoral, HTA (compression art rénale ou hypersecrétion
catécholamines)
tumeurs pelviennes : Troubles neurologiques (douleurs d’un
membre inférieur, monoplégie et/ou troubles sphinctériens)
tumeurs thoraciques, médiastinales et postérieures : signes
d’appel respiratoires banaux, Compression médullaire si
sablier : Urgence thérapeutique
tumeurs cervicales : Souvent prises pour des adénopathies,60 % SONT D’EMBLÉE MÉTASTATIQUES…
Chez l’enfant de plus d’un an ...
• métastases ostéomédullaires : AEG, douleurs osseuses,
boiterie (prises pour douleurs croissance ou attribuées à
traumatisme)
• métastases orbitaires : hématomes périorbitaires
spontanés (sd de Hutchinson) parfois associés à une
exophtalmie
Chez des enfants plus âgés...
• envahissement MO (purpura et anémie) faisant évoquer
leucémieTHÉRAPEUTIQUE
Tumeurs localisées opérables d’emblée :
chirurgie seule
Tumeurs localisées non opérables d’emblée :
chimiothérapie néoadjuvante
Formes métastatiques : formes de mauvais pronostic (30 % survie)
chimiothérapie néoadjuvante
Chirurgie
chimiothérapie d’intensification (autogreffe CSH)
ttt entretien: acide rétinoique, anti GD 2 +/- IL2
radiothérapieTHÉRAPEUTIQUE
CAS PARTICULIER DES 4S (sans amp N-Myc)
Chez le nourrisson : Sd de Pepper ou 4 S :
HMG sans anomalie biologique hépatique/hémostase.
MO
métastases sous-cutanées (nodules bleutés intradermiques)
tumeur primitive surrénalienne ++
1/3 sont susceptibles de régresser spontanément (en l’absence de
signes de détresse vitale au diagnostic : surveillance simple ;
chimiothérapie en cas de progression ou de détresse vitale)
60 à 80 % de survie à long termeCAS N°6 Gabrielle, 12 ans Tuméfaction latéro-cervicale droite, douleurs nocturnes hanche D Bilan local : TDM cervical : masse ganglionnaire d’allure tumorale TEP-scan : fixation masse cervicale droite Biopsie ganglionnaire : maladie de Hodgkin scléro-nodulaire confirmé
LYMPHOMES
12 % des tumeurs de l’enfant
maladie
maligne du système lymphatique, plus fréquente
chez immunodéprimés, secondaires après guérison LA
Maladie de Hodgkin
• 15 à 30% des lymphomes malins de l’enfant (5 à
10 % des MDH)
• 2 fois plus fréquente après 10 ans
• Traitement : chimiothérapie +/- Radiothérapie
• 90 % guérison (sans RT dans 80 % des cas)LYMPHOMES
LNH
4 types rencontrés chez l’enfant :
lymphomes de Burkitt, 50 à 60 % LNH
endémiques associés EBV : atteinte face
Sporadiques + fréquents en Europe : 90 % abdomen
lymphomes lymphoblastiques : dans résidu thymique
médiastinal
lymphomes anaplasiques à grandes cellules :10 %
Fièvre, adp inflammatoires, atteinte cutanée
Lymphomes B diffus à grandes cellules (> 15 ans)LNH
origine tumorale tissulaire :
≠ leucémies (origine médullaire : > 25 % de cellules tumorales)
Chimio-sensibles et chimio-curables :
radiothérapie d’indication exceptionnelle
pas de chirurgie
Survie globale à 5 ans > 70%CAS N°7
Thomas, 14 ans
Douleur basithoracique G lors d’un fou rire
Clinique :
palpation tuméfaction pariétale 8 cm
diminution MV base GRP : masse suspecte
Imagerie :
TDM thoracique masse suspecte arc post 10è cote G +
épanchement pleural
IRM lésion envahissant 3 dernières cotes G et parties molles
musculaires (10 x 7 x 8 cm)
Biopsie : Tumeur d’EwingTUMEURS OSSEUSES MALIGNES
Epidémiologie :
rares : 6 à 10% cancers de l’enfant
Incidence augmente avec l’âge (Pic 15 ans)
Ostéosarcome et Tumeur d’Ewing : 90%
Chondrosarcome, adamantinome, chordome et lymphomes
osseux.OSTÉOSARCOME
la plus fréquente des tumeurs malignes primitives de l’os
60 % 10 à 20 ans
SR 1,4 garçons pour une fille
Ostéosarcomes localisés
80% autour genou
Métastases au diagnostic 10 à 20% des cas
Poumons 85 à 90%
autre os (plusieurs = très mauvais pronostic)OSTÉOSARCOME
OSTÉOSARCOME
Traitement :
chimiothérapie néo-adjuvante
Chirurgie conservatrice (exérèse large) voire amputation
(envahissement local, nerveux, infection, envahissement peau)
Chimiothérapie adjuvante (selon réponse à la CT: degré de
nécrose observé dans la tumeur réséquée)
Pronostic :
Localisés : 70% survie sans évènement
Métastatique, pronostic d’autant plus réservé que multiples
atteintes osseuses ou 2 poumons : exerese chir ou
radiothérapie (peu efficace)
Récidives :
métastases pulmonaires (20-30% survie 5 ans)
métastases osseuses sont de plus mauvais pronostic.TUMEUR D’EWING seconde décennie de la vie pas de facteur environnemental identifié PNET: invasif (corticale osseuse, muscles, tendons sans en modifier structure) et extension à distance silencieuse
TUMEUR D’EWING
Formes localisées : survie 65 %
Chirurgie si localisée opérable
Chimiothérapie d’induction, chirurgie puis selon réponse
histologique : chimiothérapie, chimiothérapie HD, radiothérapie
Formes métastatiques :
chimiothérapie conventionnelle : rechutes fréquentes, 10-15%
survie
métastases pulmonaires seules : 40-50% de guérison par
consolidation = chimiothérapie HD + greffe CSH ou radiothérapie
pulmonaire à la dose tolérable
Métastases osseuses et/ou médullaires très mauvais pronosticTUMEURS MÉSENCHYMATEUSES
MALIGNES
tumeurs se développant à partir des tissus de soutien
(musculaire, conjonctif, vasculaire, nerveux ou adipeux)
reproduisant types cellulaires variés (musculaire,
fibroblastique, endothélial,…)
chimiosensibilié et pronostic différents
Exemples : léiomyosarcome, synovialosarcome, schwannome,
liposarcome…TMM : RHABDOMYOSARCOME
• la + fréquente (60 à 70%)
• 5 % tumeurs solides
• cellules musculaires striées
• Pic d‟incidence est la petite enfance avec un âge médian de 5,5 ans au
diagnostic, mais ces tumeurs peuvent aussi se rencontrer chez l‟adulte, de
façon rare
• Localisations: tête, cou, système génito-urinaire
• Facteurs pronostiques: âge < 10 ans, taille < 5 cms, site favorable ou
défavo. orbite vs par méningé, gg, histologie: embryonnaire vs alvéolaire,
qualité chir initiale
• EFS à 5 ans 31 % à 93 %, OS 42 % à 98 %
• Association : anomalies congénitales au niveau arbre génito-urinaire ou SNC, NF1,
Li-FraumeniRMS
DIAGNOSTIC : UN PROBLÈME D’OEIL
Strabisme
Exophtalmie
Reflet pupillaire à l’œil nu ou sur les photos
< 1 an : rétinoblastome +++
Lymphome, rhabdomyosarcome
Tumeur SNCDIAGNOSTIC : UN PROBLÈME D’OEIL
RÉTINOBLASTOME
tumeur maligne intraoculaire la plus fréquente
60 % unilatéraux : la plupart de ces formes non-
héréditaires
formes bilatérales ou multifocales sont héréditaires :
mutation gène RB1 a un risque supérieur à 90% (risque
accru de développer autres types de cancer)RÉTINOBLASTOME
Forme unilatérale : énucléation +/- thérapie adjuvante
Forme bilatérale : traitement conservateur pour au
moins un oeil
laser seul ou avec chimiothérapie
radiothérapie externe limitée aux tumeurs oculaires larges
infiltrant le corps vitré (sarcome secondaire)
Pronostic vital (lié au RB) excellent
Suivi à long-terme : risque nouvelle tumeur primaireAVENIR APRÈS LA MALADIE ET LE TRAITEMENT
Surveillance prolongée
Rechute
Séquelles
Fertilité, aide à la procréation
Insertion sociale/professionnelle
Second cancerMerci de votre attention.
Vous pouvez aussi lire

















































