3 millions de français concernés: les maladies rares, du malade au médicament 28 février 2020 (journée intern maladies rares)
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous

3 millions de français concernés: les maladies rares, du malade au médicament 28 février 2020 (journée intern maladies rares)
Plan de la conférence (hopefully!) • Quelques généralités: MR 3 million?, quelles maladies? génétiques et « non « génétiques » ou causalité complexe.., errance diagnostique, que peut on espérer du séquençage du génome en diagnostic • Utilité du diagnostic, nécessité de mieux connaitre chaque maladie • Du patient au médicament: quelques histoires plus ou moins personnelles • Pour les questions: Crispr (2h), dépistage en population générale (2h), autres?
3 millions de français? Cf site officiel EU Pas tous atteints en même temps, toute la vie… Incidence /prévalence, impact sur famille proche…

Les maladies rares • Plus de 6-7000 maladies rares…. • La grande majorité ont une cause génétique • Les autres: causes complexes (en général mal connues) associant prédisposition génétiques et causes environnementales (infectieuses, mode de vie, exposition à produits toxiques, ex amiante, ): ex maladies autoimmunes rares, narcolepsie, cancers rares • On découvre actuellement de nouvelles maladies génétiques chaque semaine!

Quelques maladies génétiques rares pas
si rares
• Mucoviscidose: 1/2500-3000
• Myopathie de Duchenne 1/8000 garçons
(prévalence en pop pédiatrique)
• Amyotrophie spinale 1/7000
• Drépanocytose 1/700 nouveaux nés atteints en
Ile de France (origine DOM/TOM, pays
africains)
• Retard mental avec X fragile ~1/5000 garçons,
1/7000 filles
• Maladie Huntington, Phénylcétonurie 1/12000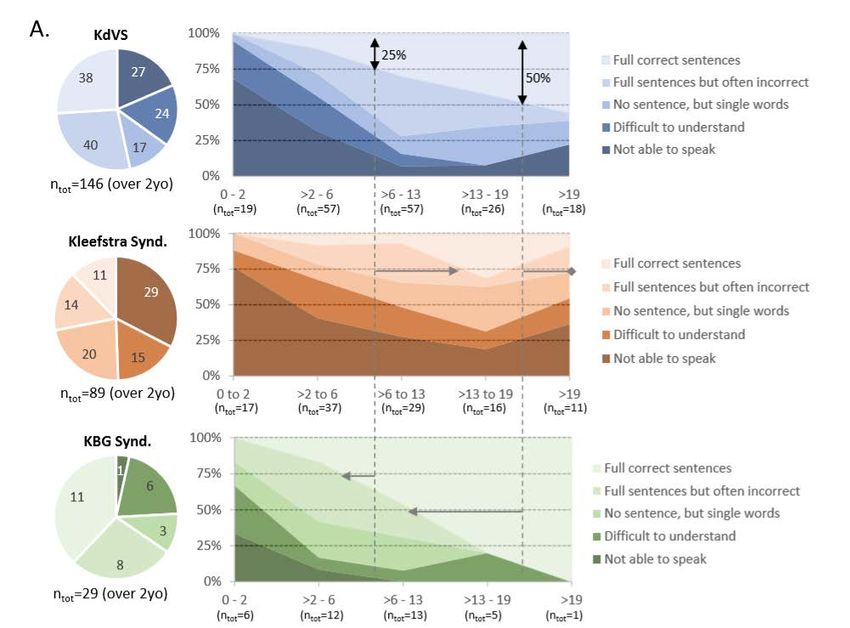
Maladies rares et maladies communes… • Des pathologies non rares peuvent être la somme de nombreuses maladies rares: exemple rétinopathies responsables de déficience vision précoce ou tardive (>100 MR, certaines avec comorbidités, ex BBS), surdité congénitale (100 MR), déficience intellectuelle avec ou sans autisme ou épilepsie (1,5-2,0% enfants et jeunes adultes, dont ~ 60% ont des causes génétiques, ~1000 MR) • Il existe des formes rares de maladies communes, en général plus sévères, début plus précoce, formes familiales: Alzheimer, Parkinson, Diabète de type 2, hypercholestérolémie, troubles du rythme cardiaque, cardiomyopathies…) sont souvent très intéressantes pour connaitre les mécanismes de maladies communes (et développer médicaments)

Les étapes de la prise en charge d’une maladie rare
• Le diagnostic, aussi précis et rapide que possible
(en fonction des connaissances du moment) pour
éviter l’errance diagnostique: donner un nom, une
cause, risque de récurrence familiale? (conseil
génétique et DPN éventuel), orienter et anticiper la
prise en charge et le traitement
• Traitement: spécifique s’il existe, symptomatique
le mieux adapté, préventif de « comorbidités »…
• Prise en charge para ou non médicale permettant
une meilleure qualité de vie, insertion scolaire et
sociale…, tenant compte des spécificités de la
maladie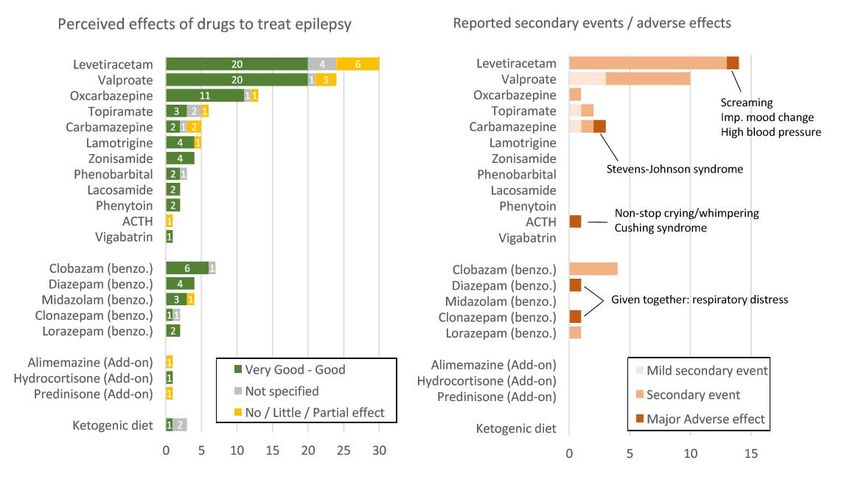
Variabilité du génome humain • Environ 6 millions de différences entre séquences de 2 humains de même sexe (~0.1% comparé à ~1.0 -1,5% entre homme et chimpanzé, dont ~8000 codantes (avec effet sur séquence de protéine codée) • Environ 60 mutations de novo entre enfant et génome de ses parents • Une « erreur /variant pathogène» d’une seule lettre (ou 2 pour maladies récessives) sur 6 milliards peut entrainer maladie sévère, par ex myopathie ou déficience intellectuelle sévère avec ou sans épilepsie ou troubles du comportement (autisme)

7681 ccatcttctc tttgtgcatg ttggtctccg tgtcccaatt tcccctttct atagggactg cagtcctaat gaattagacc ccaacaaacg acctgatatt aacttgatta cctccataaa
7801 gatcctattt ctaaataagg ccacattttt ttgagatact aggaattagg acatcaatgt atcttttatg tgacagacat ttcaacccat tagagttacc taacctccct cctaacacca
7921 cttccccttt ataaaatgag gataaaagtg ctgacctcac agggctgtgg agaacctggg gctatgcatg tagaaggatt agcacagtgc ctggcacatg gctggaaggc atcaaatgtt
8041 agctagtatt attatgaaat ggggatatag agccttagag ctcaatttat tttgctttgc ttatacagaa gtccatatgg ataacatttt cctccaactc taaagggcat aatgattttt
8161 cataacagcg taagttgatt tttacatctt gtactttaca aaggaactat atatttgaat aaaatttact ttttatttga gtattgccat gtattcatac tatgatacaa ttgccttgaa
8281 taaatacctt actcccagta agtaaataaa ccctaaatgt taaaaatctg aacaatttaa acatggctag aaaatgcacc ttctatatta ttcctaaaat aaaagaaata aaggctctaa
8401 aatgcaatat tgaattcccc caaccatgct gatgtaggta aactgtattt cagatattgg gaaatagcct cataaactga gaagaacacg gcttttagat tcaagtacat atggattcaa
8521 cttccacctt tacccctaca gctctgtgac cagtgggaag ttatgtagct ttgttcagcc ttggtttctt catctgcgaa attaggaaaa taatactcct tcaaaagtga gagagcgtaa
8641 cctgcagtgg atgaatggat aaacaaaatg tgctatgtac atataatgga atattattca gccttaaaaa ggaaggaaat tctgacacac gctataacat ggattaacct tgagaacatt
8761 atgctaagtg aaataaacca gtcatcaata gacagatact gtattattcc acttatgtga ggtacctaaa gtcatcaaat tcatagaggt aaaaaacata aaggcttttg ccaaggtcca
8881 gggggagggt agaatgagga gctactgttc actgggtaga gagctttaat tttgcaagat gaaaagagtt ctggaaatgg ctgctcatga tggtttcaaa acgatgtgaa tatccctaat
9001 gccactaaac tgtacacttt aaaaatggtt acgatggcaa atcttatgtt atatatattt tactctaatt tttaaaataa atttaaaaaa taaatcctaa aacattgttt aaaatgtgaa
9121 atagtttaag gaaatcccct aatgtgacgc ctagaataga agcaggtagc tattagacac attccttctt tagttttctc cctcctcccc accataaata gttgcaaaaa taattggagt
9241 gtgcacatag ccaaagattt aatgccacat agccaaacaa acaccagata attcaggaac ccttgattct gaagtgaagc ttataagaag atgaaccaca ttggatcagg aaataagaaa
9361 accagtccat atgttgcaat taacttgttc tgtgattggg agcaagtcac ttagcttctt tggacctgtt ttctcatctg tcaaataagg agggttgaac taggtaatct aaatgaaatc
9481 caagtcctta gaggcttgta ttacataaat caagtcaaga catggtattt aagaatgaag ggtcatagtt tagcatgcta ataattcttc ttcatgcaaa aacataggag ggggaaataa
9601 atatctttta tcgtaatacc atgataaatt tgctgggtgg ggggaggaat agattataag acaggccgaa aggagcaatt aatagcgaaa tgtcacacta ttctatatca aatgttatgc
9721 atttaaaaga atatgtcagt tttgcaagat gaacaagttc tagaaatgtg ttgcacaatg atgtgtatgt agttaacaac tctgcactgt acatttaaaa tggtgaagat ggtaaatttt
9841 atattatggg ttttttgcca caacttaaaa aaagaatatg ggcaactatt ttctttcttt aacatcctca tttttcaaaa acaataccag tggttttcaa gcttttttac aaagagcaaa
9961 tccattcttc taacaaagtt tcataagaaa aataactgta aaaaaaaaat atggagttga aagtgaggca tgagatggag gtaacgaata ttcccagtat gagcctctct cccttcttgt
10081 tccttgggcc tgctcctgag ttctgcaaag actccctagt gctccaggag gctggtttaa aaatcaatgc tttatactct acaaagaaat gtaggccagg cacaatggct cacacctgta
10201 atcccagcac tttgggaggc caaggtgggg agatcacttg aggtcaggag ttcaagacca gcctggccaa catggcgaaa ctctatctgt acaaaaaaca ccaaaattag ccaggcatga
10321 tggcgcatgc ctgtagtccc agctactcat gaggctgagg tgggaggaac gcttgagccc aggaagttga ggcggcattg agctgagata gcaccactgc actccagcct gggtgacaga
10441 gcaaggccct atctcaaaaa aaaaaaaaaa aaaagaaaag aaatgtaaat ggccctcacc atgcattcga ctgggaatta atggtggtag agctttgttc aactgagccc cacaatccat
Génome humain = 2x500 000 pages comme celle-ci
10561
10681
10801
ctaaccattt
ataaaaatgt
taacatcaat
actgcaatac
gtataaatga
tatattccag
tgtaaaagtg
ctgaattcat
attattgtta
gaccttagag
gaagggaccc
agcacaggga
ggggtatttc
aagctccagg
ataaaaacat
tatctgcttt
aaaagcatag
gaatcagata
caggaagaga
caggagaagt
cagtccctac
tattatggat
attttattaa
cctcaaggaa
atactagagg
tctatccatt
ctcactctct
taaaaaaccc
cattcattta
ggtacaagag
ccaagtttag
tttattcgaa
acatgtttca
cacattctca
agcatttctt
aaaggtcatt
10921 gtaaagaaat gtggaaaagg caagaattat gcacagaggg ctatgggagt accaagccag ctgtgaggga gccagggaag acttcttgaa ggaaggatta agagctagct gctcaatgac
11041 aggagaaggt catggaaggt gtagacaaaa taatgagcat gtgtccatgt gtgggaagtg gcttggtgtg ttgggaagaa cacaacacag agaaggaatg ggagacatga agccacaggc
11161 agggttgtgg agagttttgc atgtgagtag ggcctgtctg catttttctc ccttggcttt gcctggctcc tttccttcca tcccagggga cctccccctg acactccaca gagctttgag
11281 ttctataatc tgtagcttat tcccactcca tggagaaaga ggaaagaagg ctaagaggaa gaaggaaagg gcatttcacc tcctcagtgg aagctaccat agaaagtcag atcaggccgg
11401 gcacggtggc tcatgcctgt aatcccagcg ctttgagagg ctgcggctgg cggatcacct gaggtcagga gtttgagacc agcctgggca acatggtgag accccgtctc tactaaaaat
11521 acaaaaatta gccaggcatg gtgctgtgtg cccgtagtcc cagctactgt ggaggctgag gcaggagaat tgcttgaacc caggaggcag aggttgcagt gagccaagat agtgccaccg
11641 cactccagcc tgggcaacag agcgaaaact ctatctcaaa aaaaaaaaaa aaaaaaaaaa aaaagatcac aacgtttacc cataaaagaa aacaacaatg ttgcttcatg agtccttgat
11761 gggtttctga gaggcagaag catttgacct gaaggtgctg tgtggaaagg gcccgctgag aactcctcct ccaccaactc cccaggacca agctatctat aggtgctggg tgttcacggc
11881 tgacttgccc agcatcaggg aggcctggtc cctcaacctc agttcaaggc cctcaggtac ttggagctca agacttcccc tccttaggac actctacttt ccagctttgt ttaatgaaaa
12001 cattcacttc tgattcaata gcattagaaa acccagattt catttccctt tacaacagca tgaacacctg aatcgttccc ctactgggag tctctttgga gtttgaatca aaatgctgac
12121 cataatggag ggacttctct ctcttatatg aacatccact agataaacat caaacaaacg tagatatgaa aatgctgaca tggggcagac aggaatgaaa atctgttagg aaagacctca
12241 gaattcccta tactctttct aagtagatag agatacagat aaacatccca aattttaagt ttttaaatct tttagttgat aaaaatcaaa gcagtgactg atcccatgtg gtccagacca
12361 ctgttctcat ctaaggcaac ctcagaaacc caacagcccc attgagtaga tttagaccac aggttactgg gagaagatgg atttcaggaa cagcaaaaat caaaagcaaa tattcttttg
12481 aagcttgcaa aatcactgtt ttagaatctg aattatattg gcaaccagag agaatcaagc attcttcttc atttcccaaa atactaacat ttcccttgtg ggaactgatt ctcgatttct
12601 atttattaca aaaggaagaa aaacatgttg aggtcttaca ttctcctatt ttttttcact tttccattgt cctgaatagc aaagtaccat attactcaga atatgatttt gcatatgcta
12721 tcagataaat ctaaaacatt tttgtggcta aatttagatg tttgtataac atactggggg aaaatcactt aagtgatttt ggcactaata ctcaaaagac gtgtttctac actttcagta
12841 aaactacaac aaacgtgtta aatatttggt aaaaacaggg aggcatggtc aaaatttgaa aatcaaaaag tcttaaatga agaaagataa gatgatggct ctacattccc atgaactagt
12961 acaacaatta ttttttttaa aacatacata tactgtttct agagcagttt tgggttcact acgaaattaa gcagaaaata tagaatttcc atataaccat cccacaccat gcgtagcctc
13081 cctgatcaag atccctcacc agatggcacg tttgttacaa ctgattaacc tacattgata catccttagc acccacaggc catagttgaa ttagggttca ctacagtcta tgggtcttga
13201 caaatgtgta atgccatgta gccaccatta taacatcaca cggaatagtg tcattgtcct aaaaatcccc tgtgctcaac ttgttagtcc ttccttctcc ccagttctgg acaaccactg
13321 atctttttcc tgtctccata attttgactt ttccagaatg tcataaagtt ggaatcacgc agtatatagc cttttgagat gggcttcttt cacttaataa tatgcattta atgttcttcc
13441 ataatttttc actgtctaga tataccacag tttatccatt cacctactga agaagagctt ggttgcctcc aagttcaaca atttgctttt aaacttactt atttaaagag tggctagctc
13561 agaataattg ctgtctttat tttcttattt aatttcaaag gtgaccaaga caactcttaa ggaacgcatg aattcccctt agcaacaact ccaacagact gtctgacttt gcttgcttct
Les étapes de la recherche sur les
maladies rares: 1) diagnostic
• Identifier les gènes dont les mutations sont responsables
d’une présentation clinique plus ou moins
précise/spécifique. Savoir quels variants dans ces gènes
sont pathogènes, associés à formes sévères ou moins
sévères ou avec comorbidité particulière, ou bénins
• Pour les maladies probablement non génétiques: identifier
des biomarqueurs pour le diagnostic spécifique, tests
sanguins (métaboliques, immunologiques), imagerie (IRM,
avec marqueur spécifique, anapath), signes cliniquesLes étapes de la recherche sur les maladies rares: 2) connaissance clinique • Son évolution au cours du temps (histoire naturelle), variabilité d’expression clinique, pénétrance, comorbidités (ex ataxie de Friedreich), quels handicaps, • Réponse aux traitements symptomatiques (essai clinique si possible, ou au moins étude de cohortes de patients) • Sa fréquence (épidémiologie) dans diverses populations, environnements… (ex Minimata) • Une approche participative: les patients et leur famille comme acteurs du recueil de données: projet (international) GenIDA sur (à terme) les > 1000 formes génétiques de déficience intellectuelle avec ou sans autisme ou épilepsie
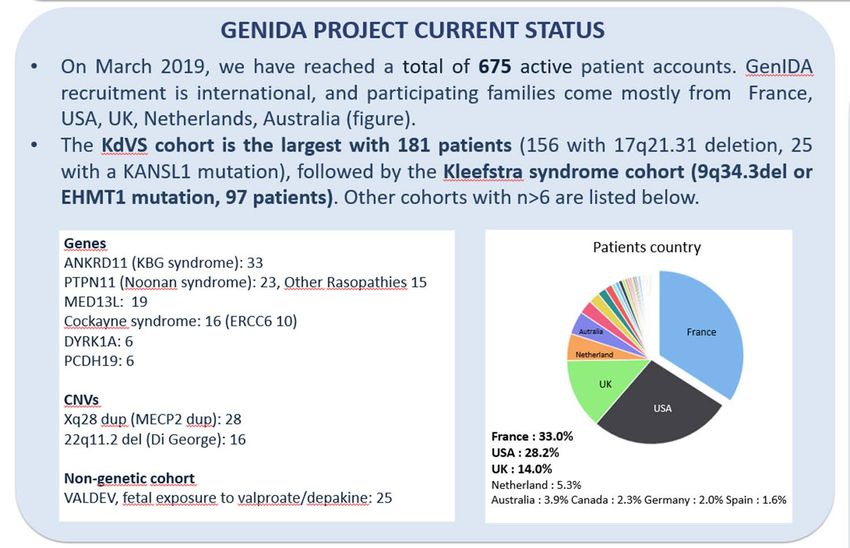
https://genida.unistra.fr March 2019
June 25 2018: 492 filled questionnaires
June 4 2019: 722 filled questionnaires
50 % yearly growth (fevr 2020 815)
Two major cohorts:
Koolen de Vries syndrome 202
Kleefstra syndrome 101
France 33.9%
Netherlands:
4.7%
UK:
14.1%
USA: 26.4%Les étapes de la recherche sur les
maladies rares: 3) mécanismes
pathologiques
• Quelle est la fonction du gène, comment la dysfonction
conduit elle aux dysfonctions physiologiques: biochimie
et biologie moléculaire, physiologie sur le patient, ou sur
des modèles animaux (souris génétiquement modifiées le
plus souvent, mais aussi rat, poisson zèbre, drosophile,
crapaud xénope), modèles cellulaires (dont cellules
souches pluripotentes induites iPSC, organoïdes)
• Comment peut on corriger certaines de ces dysfonctions
physiologiques sur ces modèles: recherche initiale de
stratégies thérapeutiques
• Cribles de molécules potentiellement thérapeutiques (ex
iSTEM/AFM)Les étapes de la recherche sur les
maladies rares: 3) recherche
thérapeutique préclinique et clinique
• Tester quelques stratégies thérapeutiques prometteuses
(petite molécules, ou thérapie génique, ou anticorps
monoclonaux, ou chirurgie…) sur les modèles les mieux
adaptés
• Convaincre un labo pharmaceutique ou biotheque (ou créer
une start-up) pour développer approche thérapeutique qui
semble la plus prometteuse, essais de toxicité… vers essais
cliniques phase 1 ou 2 (si molécule en repositionnement)Les étapes de la recherche sur les maladies rares: 4) connaitre impact de la maladie sur qualité de vie patient/famille • Recherche clinique et Sciences humaines et sociales: économie, psychologie, sociologie et anthropologie, pour connaitre et proposer des interventions adaptées..
le séquençage du génome va-t-il résoudre
le problème de l’errance diagnostique?
• Séquençage à très haut débit permet de développer plus
systématiquement le diagnostic des maladies génétiques, y
compris pour celles qui sont très hétérogènes (surdité congénitale,
rétinite pigmentaire, retard mental monogénique…)
• Errance diagnostique: le temps (et nombre de consultations et
examens divers) entre la constatation des premiers symptômes
inquiétants (par les parents) et le diagnostic précis: de quelle
maladie s’agit il et quelle en est la cause?
• Il existe (au moins) deux types de diagnostic: le diagnostic
(bio)clinique, par ex rétinite pigmentaire, myopathie des
ceintures, syndrome de Bardet Biedl, épilepsie et déficience
intellectuelle… et le diagnostic de la cause: quel gène, quelle(s)
mutation(s) chez le patient, pour les maladies monogéniques,
quelle CNV (copy number variant) ou quel type précis (quelle
dysfonction) pour une maladie (apparemment) non monogénique
16Diagnostic et séquençage (panel,
exome, génome..)
• En principe, si c’est génétique, on devrait trouver la
réponse par le séquençage du génome du patient (en le
comparant à celui des parents non atteints, séquençage en
trio très utile pour l’interprétation)
• On ne sait si génétique que 1) si tableau clinique très
spécifique (c’est rare) ou 2) si familial (rare, et
environnement aussi familial) ou 3) quand on a trouvé un
variant pathogène dans un gène adéquat
• Mais difficile de prouver ou éliminer des causes
génétiques liées à des mutations dans régions non
codantes, ou mécanismes plus complexes, oligogénisme,
implication de variants hypomorphes (ex syndrome TAR)
11/03/2020 17Causes de l’errance • Temps entre première constatation par les parents et prise au sérieux par le 1er médecin (généraliste, pédiatre, spécialiste de ville) puis jusqu’à doute: ce pourrait être une maladie rare (génétique ou non) et adressé à spécialiste de préférence centre de référence ou de compétence et temps obtention consultation Améliorable par une meilleure formation et information des médecins de divers niveaux, information fiable sur Internet pour professionnels et familles (Recherche SHS pour évaluer efficacité des diverses approches?) • Avoir plus de spécialistes et de conseillers en génétique 11/03/2020 18
Causes de l’errance (2) • De la consultation par spécialiste au test de séquençage adapté • Avant 2000, nombre de maladies diagnosticables était limité. 2000-2012 test gène par gène, souvent long, pas efficace et couteux, on s’arrêtait après 2 ou 3 gènes au maximum (et grands gènes non séquencés) • Après 2012: développement progressif de séquençage haut débit: panel de gènes connus (au temps t) pour la présentation clinique, ou exome = toutes les régions codant pour des protéines (2% du génome) • A fait énormément progresser le diagnostic mais problème d’accessibilité (inégalité territoriale), et des connaissances qui continuent à progresser, chaque semaine de nouveaux gènes de maladies identifiés • Techniques s’améliorent: un exome de 2012 rate 20% des séquences codantes. Le génome complet permet un exome complet 11/03/2020 19
Comment interprêter • Plus facile pour certains gènes (accumulation de données publiées, de bases de données spécifiques, type de mutations causales) que pour d’autres, pour certaines mutations (par ex mutations troncantes = perte de fonction) que pour d’autres… • Utilisation de logiciels de prédiction bioinformatique (il en existe beaucoup, pas toujours d’accord entre eux..) indicatifs mais on ne peut pas se baser entièrement sur eux • Tests fonctionnels (recherche) ont pu établir caractère pathogène ou non pour certains gènes et certains variants • Mais…… les VUS les VUS les VUS 11/03/2020 20
Le problème des VUS/VSI
(variants of uncertain significance ou Variants de signification incertaine)
• Pour la plupart des gènes, on a des variants dans
séquences codantes qu’on ne peut interpréter comme
bénins ou pathogènes avec outils bioinfo ou bases de
données (individus controles et atteints)
• Dans certains gènes, plus de VUS que de variants
pathogènes. Problème d’autant plus important que gène
découvert récemment ou maladie très rare (peu de
données dans bases de données)
• Insuffisance des bases de données, par ex Clinvar,
faible contribution des labos français (pas le temps…), et
il faudrait des bases de données avec plus d’info sur
phénotype. Considérations éthiques trop stringentes
entrainent perte de chances pour les patients
11/03/2020 21Le problème des VUS/VSI
(variants of uncertain significance ou Variants de signification incertaine)
• Problème encore bien pire si on considère séquences
non codantes (> 98% du génome)
• Ex TAR syndrome (thrombocytopenia absent radius)
• Comment faire?: intelligence artificielle? mais il faut
beaucoup plus de données (mondiales) de qualité,
OMICS (transcriptomique ciblée commence à être
utilisée au cas par cas dans des labos diag, mais pas de
manière généralisée), métabolomique...., modèles
cellulaires ou animaux pas trop chers (zebra fish) mais
comment les appliquer à échelle suffisante?
• Donc il faut encourager (financièrement) les labos
diagnostics à entrer des données dans bases
variants/phénotype (même peu précis)
11/03/2020 22La route difficile des thérapies spécifiques • Une première thérapie spécifique (2011) pour… ~3% des patients atteints de mucoviscidose: ivacaftor (Kalydeco) pour les patients avec au moins une mutation G551D (prix >150 000/an). Pour les porteurs de mutation la plus fréquente DF508, combinaison ivacaftor-lumacaftor (Orkambi) a AMM depuis 2016 en dépit d’effet thérapeutique plus modeste et prix très élevé, à partir de 12 ans (1500 patients en France) et Trikafta en 2019 beaucoup + efficace (300 000$) • Thérapeutiques pharmaco basées sur modèle animal: losartan et maladie de Marfan?, échec Syndrome X fragile • Rarement, connaissance de fonction entraine mise au point de traitement diététique à effet important: exemple classique phénylcétonurie (depuis année 1960!), plus récemment déficit en transporteur neuronal de glucose Glut1 (régime cétogène), et beaucoup plus récent, déficit en transporteur riboflavine (vitamine B2)
Traiter: molécules nouvelles ou
nouvelles applications
Molécules repositionnées (repurposed) peuvent être identifiées
sur la base de hypothèses mécanistiques, ou par criblage haut débit
de molécules dans cellules (par ex iPS dérivées de patients, iStem)
Teachey et al. 2009
Kurolap et al. N Engl J Med 2017 off-label compassionate therapy for three
patients, complete remission
Nicotine patches very efficient for a rare genetic form of
epilepsy!
Brodtkorb, E and Picard, F 2006Quitterie Venot ….Fabiola Terzi &
Guillaume Canaud (Necker Enfants
Malades) Nature june 2018
Syndrome de Cloves, un syndrome sporadique que l’on pensait
ultrarare (décrit 1ere fois en 2007 sur 7 patients, différent de
Proteus Syndr/Elephant man), avant que l’on s’aperçoive (Kurek et
al. AJHG 2012) qu’il est lié à des mutations « gain de fonction »
somatiques (donc de novo) en mosaïque dans le gène PIK3CA,
aussi presentes dans d’autres PIK3CA-related overgrowth
syndromes (PROS) défini en 2015
CLOVES syndrome (congenital lipomatous overgrowth, vascular
malformations, epidermal naevi, scoliosis/skeletal and spinal
syndrome) is a genetic disorder that results from somatic, mosaic
gain-of-function mutations of the PIK3CA gene (a regulator of
cell growth via mTOR pathway), and belongs to the spectrum of
PIK3CA-related overgrowth syndromes (PROS). This rare
condition has no specific treatment and a poor survival rateQ. Venot….
Guillaume
Canaud (Necker
Nature juin 2018 Enfants Malades)
we describe a postnatal mouse model of PROS/CLOVES that
partially recapitulates the human disease, and demonstrate the
efficacy of BYL719, an inhibitor of PIK3CA, in preventing
and improving organ dysfunction. On the basis of these
results, we used BYL719 to treat nineteen patients with
PROS. The drug improved the disease symptoms in all
patients. Previously intractable vascular tumours became
smaller, congestive heart failure was improved,
hemihypertrophy was reduced, and scoliosis was attenuated.
The treatment was not associated with any substantial side
effects.De l’exome à un traitement: 3 histoires
strasbourgeoises
• Enfant avec crises fréquentes d’épilepsie nocturne pharmacorésistante:
atteinte sommeil, comportement, cognitive, déscolarisation: exome révèle une
mutation dans un gène codant pour sous unité de récepteur nicotinique à
l’acetycholine, 3 articles suggèrent que pourrait répondre à nicotine: effet
bénéfique +++ de patch de nicotine sur l’épilepsie, sommeil et comportement;
enfant est rescolarisé (Anne de St Martin, J. Chelly)
• Un cas de trouble des mouvements (dystonie): exome révèle mutation dans
gène ADCY5 (adenylcyclase), une publi française de 2019 indique que pourrait
répondre à caféine… confirmé chez le patient (A de St Martin, J Chelly)
• Homme 17 ans Une forme atypique de myopathie des ceintures à évolution
progressive débutant à 2 ans: pas de traitement, Odyssée diagnostique :
Investigations dans 3 centres de maladies neuromusculaire de référence: Paris
Necker, Ankara, Strasbourg: analyse panel de 140 gènes (V Biancalana) :
myasthénie liée au gène DOK7: traitement efficace salbutamolAmyotrophie spinale: du gène au traitement
• 1990-95 de la localisation à l’identification du gène SMN1 (et sa copie
partiellement active SMN2 (Judith Melki, Paris)
• 1997-2000 Rôle du nombre de copies SMN2 dans la sévérité
• 2009 Travaux publiés de thérapie génique préclinique sur modèle souris,
vecteur AAV9 (M Barkats, Généthon, B. Kaspar USA)
• Création AveXis en 2015 (avec license de brevet Genethon) par B
Kaspar, développement en vue essai clinique, rachat par Novartis en
2018 8,7 milliards $, mise sur le marché en 2019 au prix de 2,1 million $
par injection (unique)
• En parallèle 2012-17: après travaux académiques (A. Krainer, CSHL),
développement thérapeutique d’oligo antisens pour corriger épissage
SMN2 (Ionis, puis repris par Biogen, nusinersen), premiers essais
cliniques publiés en 2016 et approuvés par FDA dec 2016 Prix 750 000$
première année, 375 000/an après
• Mais traitements efficaces que si administrés avant début des
symptômes: donc soit pour 2eme nouveau-né atteint dans famille, ou…
dépistage néonatal systématiqueLa position incompréhensible de Mme Buzyn sur le dépistage néonatal de l’amyotrophie spinale • La loi actuelle empêche de faire un test génétique (cad sur ADN) en dépistage. L’AFM Téléthon a proposé un amendement pour permettre un tel dépistage • réponse catégorique de la ministre: un test génétique fait courir le risque « du tout génétique, de la génétique qui voudrait tout régler et tout prévoir » amendement rejeté • Pendant ce temps… • Allemagne (Westphalie et Bavière), 213 000 tests en 18 mois, 30 patients identifés, lors 1ere année, 10 patients avec 2 or 3 copies SMN2 copies traités par Nusinersen, débutant 15–39 j après la naissance, 7/10 patients avant début des symptomes; Australie 104 000 tests en un an (aout 2018-juil 2019), 9 patients traités de 16-37j apres naissance, Belgique 17 000/an, 3€/test
Une histoire personnelle, la myopathie myotubulaire (centronucléaire) • Petit début collab 1992-93 • Début réel : une patiente particulière permet localisation beaucoup plus précise • Un étudiant en thèse, J Laporte: identification du gène en 1996 Nat Genet • J Laporte continue comme postdoc, puis CR puis DR.. Fonction du gène (au départ modèle levure, puis cellules en culture, puis souris). Découverte d’autres gènes impliqués dans des maladies centronucléaires montrant interactions fonctionnelles. Donne idée de cible thérapeutique par oligonucléotide antisens, validée chez la souris. Création de start-up: Dynacure
Une autre histoire, la myopathie
myotubulaire (centronucléaire)
• 1998 Une postdoc Anna Buj Bello, initie modèle souris qui
reproduit la pathologie, premier test thérapie génique sur ce
modèle
• Part au Généthon pour poursuivre recherche
thérapie génique par vecteur AAV. Résultats
spectaculaires sur modèle souris, puis sur modèle
naturel de chien (dont mutation avait été identifiée
par J. Boehm et J Laporte). Construction de vecteur
AAV pour essai clinique chez l’homme
• Accord start-up USA Audentes – AFM Téléthon pour essai
clinique aux US principalement 12 enfants traités, résultats
spectaculaires avec correction de parametres majeurs (temps sous
respiration assistée). 2019 Audentes racheté par un labo japonais
Astella pour 3 milliards $ !Des histoires personnelles… (au départ) • Syndrome de retard mental avec X fragile 1983 un outil (sonde polymorphique), une rencontre Jean François Mattei, généticien clinicien, futur ministre de la santé, et Geneviève Mattei, cytogénéticienne , une grande famille X fra, une publication dans Nature puis d’autres collaborations (Paris, Lille, Nancy, Leiden, Adelaide, Hawaï… 1991 Identification d’un mécanisme de mutation, expansion instable de répétitions avec modification épigénétique (Strasbourg) et identification du gène FMR1 (Rotterdam); applications diagnostiques, travaux sur la mutation 1993 début des travaux sur la protéine FMRP, Barbara Bardoni, puis en 2000 sur interaction avec ARNm, Hervé Moine
Des espoirs thérapeutiques • A partir de 2002, des travaux (US, Pays Bas) sur modèle souris et drosophile suggèrent cibles thérapeutiques récepteurs mGluR ou GABA • 2008-10: Intérêt des big pharmas et de start-ups et début d’essais cliniques randomisés • Novartis et Roche: inhibiteurs récepteur mGluR5 (AFQ056, RO491752) • Seaside Therapeutics (M. Bear, soutenu par Roche): agoniste récepteur GABA-B (STX 209 = arbaclofen) • NB: correspond à période où X fragile devient “l’exemple” de pathologie autistique génétique
Promise Seen in Drug for Retardation Syndrome April 29, 2010 After learning that their son, Andy, had fragile X syndrome, Katie Clapp and her husband, Dr. Michael Tranfaglia, started the Fraxa Research Foundation « Just three years ago, I would have said that mental retardation is a disability needing rehab, not a disorder needing medication » said Dr Tomas Insel, director of the National Institute of Mental Health, who was told of Novartis results. « Any positive results from clinical trials will be amazingly hopeful »
Sci Transl Med. 2011 Jan 5;3(64):64ra1
The First Drug that Could Ease Social Withdrawal in Autism
Sept. 20, 2012
An Experimental Drug’s Bitter End
Holly Usrey-Roos, right, with Parker,
14, and Allison, 10. Both have fragile X
syndrome. Ms. Usrey-Roos said Parker
was helped by the drug arbaclofen.
By ANDREW POLLACK
Published: June 6, 2013
Now, however, the drug is being taken away. “It has not met the goals set for it in
clinical trials testing it as a treatment for either autism or fragile X syndrome” (Roche
position on the decision not to exercise the option for Seaside Therapeutics' compound
arbaclofen) . And Seaside Therapeutics, the company developing it, is running out of
money and says it can no longer afford to supply the drug to former participants in its trials.
The setback is a blow in the effort to treat autism since the drug, arbaclofen, was one of
the furthest along in clinical trials. And the company’s decision has caused both
heartbreak and outrage among some parents.
Until recently, Seaside, one of the few companies pursuing autism drugs, was considered a shining light by family
members of those with the condition. The company, in Cambridge, Mass., grew out of the research of Mark F. Bear, a
neuroscience professor at the M.I.T.April 24, 2014: Novartis Clinical September 10, 2014: Roche
Trials in Fragile X Ended abandons another Fragile X R&D
program after PhII trials flunk out
Novartis has announced that the company will
be discontinuing its development program in A long, rough path in Fragile X syndrome
Fragile X for its lead mGluR5 antagonist, drug R&D just got longer and rougher.
mavoglurant (AFQ056), following negative Roche has notified patient groups that
results in a large international clinical trial in both of its mid-stage studies for RG7090-
adults (reported in the Fall of 2013) and most -an mGluR5 therapy--failed to hit the
recently, in a trial in adolescents. In both primary and secondary goals, prompting
placebo-controlled trials, patients taking the pharma giant to shut down the
mavoglurant did not show improvement over program.
placebo in any outcome measures. "Unfortunately, Fragxis (one of the
Novartis has also announced that the clinical trials which enrolled 183 adults
current open-label extension phase of the and adolescents) did not demonstrate the
trial will be closed, hoped for efficacy based on the primary
and secondary endpoints employed,"
La question de la pertinence des tests Roche neurosciences chief Luca
utilisés (échelles comportementales) très Santarelli told the patient groups in a
sensibles à l’effet placebo(A.Curie, V. letter sent out on Tuesday.
desPortes et al PlosOne 2016). Des tests
cognitifs objectifs sont nécessairesL’adrénoleucodystrophie • Maladie démyélinisante liée au chr X, incidence 1/20-30 000 • Insuffisance corticosurrénale (maladie d’Addison) généralement présente chez les mâles, souvent le premier signe • Forme infantile cérébrale (CALD) la plus sévère, entre 5-12 ans (35-40% des garçons mutés), décès en qq années en général • Forme adulte périphérique AdrénoMyeloNeuropathie (AMN), vers 20-30 ans ou mixte (avec survenue atteinte cérébrale dans 15-25% des cas). AMN très fréquente chez femmes vectrices après 40 ans (diag. différentiel avec sclérose en plaques) • Diagnostic biochimique: niveau très élevé d’acides gras saturés à très longue chaine C26:0 dans le plasma (Moser H et al.1981), suggérait déficit du catabolisme peroxisomal de ces acides gras.(Singh, Moser et al. 1984) • Les diverses formes peuvent coexister chez les mâles d’une même famille
1ère publi à Strasbourg, en
collab. avec les Boué et
Hugo Moser:
1er contact en 1986 avec un jeune neuropédiatre parisien spécialiste de
la maladie, en postdoc chez Hugo Moser
We are grateful to Dr J-L Mandel
for the gift of St14 plasmid,
A son retour, des US, une collaboration est établie en vue de cloner le gène de l’ALD
Patrick Aubourg montre sur un cas, l’intérêt thérapeutique de la transplantation de
moelle dans l’ALD, dont
l’efficacité sera confirmée
dans les études ultérieuresIn Moser’s lab, suggestion that color vision genes (RedCP, GreenCP may be close to the ALD gene Nous décidons d’explorer cette hypothèse en marchant sur le génome à partir des gènes de vision des couleurs
1 er
thésard
Robert
Feil
Un seul patient avec ALD et déficit de vision des couleurs (de chez H Moser)
montre un profil anormal, et un réarrangement complexe sur le chr X
Claude
Travail poursuivi par 2 étudiants en thèse… SardeNATURE 361: 726-730 FEB 25 1993 Look at the title! Imposed by the Nature editor
Hugo Moser sir Peter Ustinov
(1924-2007) (1921-2004)
Times Cited: 885 Peter Ustinov portrayed Hugo Moser, the ALD
specialist, in Lorenzo’s oil. Both had Mittel-
Europa origins (German-Austrian for HM,
Russian-German for PU), the same age, the same
look, and the same accent
The paper
was out in
Nature the
week of the
London
premiere of
LorenzoEt études à Strasbourg, puis
Barcelone, sur un modèle
souris (Aurora Pujol)
Patrick Aubourg se lance sur la thérapie génique ex vivo de l’ALD, d’abord avec un
vecteur rétroviral, mais pas assez efficace, puis vecteur lentiviral dérivé du HIV
Il publie, 16 ans après identification du gène , avec Nathalie Cartier (neuropédiatre, et
actuellement présidente du conseil scientifique de la FMR), la première thérapie
génique sur l’ALD, (et 1ere utilisant un vecteur lentiviral)
Résultats positifs chez 2 patients,
et un 3ème publié un peu plus tard
920 citations Web of SciN Engl J Med 2017;377:1630-8. 8 ans pour qu’un 2eme essai clinique soit publié, sur 17 patients: résultats favorables sur 15 patients (1er patient traité en 2013) Avec une start-up BlueBird qui avait obtenu en 2012 Orphan Drug Designation pour cette thérapie
Sept 2019: 32 patients traités dans essai clinique, avec comparaison avec transplantation Cell souches hématopiétiques hétérologues
Lorenzo oil and Augusto Odone,
father of Lorenzo
Un effet spectaculaire sur le paramètre biochimique
diagnostic, mais après une première publication qui semble
montrer un effet clinique (diminuerait probabilité de
passage à forme cérébrale.. en traitement
présymptomatique), mais controversé et non confirmé.Lorenzo oil: Effet préventif sur forme cérébrale?
Brain Pathology (2010) 845–856
Lorenzo’s oil teaches a lesson about how raising early hope in
patients creates ethical issues that prevent double-blind studies,
which in combination with the lack of knowledge concerning the
molecular mechanism of a disease and the relatively low patient
numbers, have led to a situation where the efficacy of a therapy is
still unknown even after 20 years of use in probably more than
500 patients.GenIDA
Aug 2019
Treatment of epilepsy in KdVSComparaison de 3 syndromes (KdVS, Kleefstra, KBG/ANKRD11):
langageComparaison de 3 syndromes (KdVS, Kleefstra, KBG/ANKRD11):
langageComparaison de 3 syndromes (KdVS, Kleefstra, KBG/ANKRD11): comportement
July 2018
Unexpected events in the context of the KdVS
https://genida.unistra.fr
Pneumonia and asthma example: There was no question on
respiratory infections/pneumonia: parents reported in open text
Asthma Pneumonia
m 2.0 RSV virus turned into bronchiolitis/pneumonia and hospitalized for a m 2.0 RSV virus turned into bronchiolitis/pneumonia and hospitalized for a
week at 3 months old. Dx Restrictive Airway Disease (asthma for babies) week at 3 months old. Dx Restrictive Airway Disease (asthma for babies)
f 6.5 ASTHMA AGE 1 f 6.9 Laryngomalacia (0-2 years old) Croup/bronchitis/pneumonia (0-ongoing in
m 8.0 He had asthma triggered by respiratory infections from birth to around 9 2018)
years old. This subsided as he got older and is now completely gone. f 7.7 1-5 years old: Immune problems. She seemed to catch every sickness she
m 10.4 … asthma (infant), repeated pneumonias, Otitis media (birth), … was exposed to, and had recurring bouts of pneumonia, a few which ended up
m 12.9 4. Pulmonology Asthma w/ acute exacerbation, Obstructive Sleep with her in the hospital.
Apnea and re-occuring bacterial and viral Pneumonia m 8.0 Many bouts of aspiration pneumonia. Always hospitalised for severe
f 17.0 Outgrew asthma by age 5 croup requiring nebuliser adrenaline and other medications
f 18.2 Fl a de l'épilepsie, des problèmes respiratoires (bronchites, laryngites, m 8.0 Repetitive pneumonia
pneumonie, asthme) f 8.3 Last year a pneumonia with hospitalization. With six years of age.
m 27.4 Tumor behind eye age 3. Premature birth. Low birth weight. Jaundice at f 10.0 Until the age of 6 she had several pneumonias and bronchial problems.
birth. Allergies. . Lazy eye. Asthma. Hashimoto disease She has recovered and is without problems now
m 10.4 … asthma (infant), repeated pneumonias, Otitis media (birth), …
m 29.4 Ages 2-9 - severe asthma with 2x daily home breathing treatments, m 11.0 re-occuring Pneumonia
several hospitalizations m 12.9 4. Pulmonology Asthma w/ acute exacerbation, Obstructive Sleep
f 35.3 Asthma, chronic bronchiolitis, chronic croup (in other words, she is often Apnea and re-occuring bacterial and viral Pneumonia
f 18.2 Fl a de l'épilepsie, des problèmes respiratoires (bronchites, laryngites,
struggling to breathe)
pneumonie, asthme)
f 24.4 RECURRING COLDS AND CHEST INFECTIONS ONE OF WHICH LED TO
PNEUMONIA UNTIL APPROX 10 YEARS OLD
- We will add in the general questionnaire a question on respiratory track infections
- NB: same (previously known) observation on such repetitive infections for MECP2duplicationMouse models to develop gene therapy • Friedreich ataxia et myotubular myopathy1(frataxin and myotubularin genes, 1996) • Very encouraging reclinical studies in mouse (H. Puccio for FRDA, A. Buj Bello, J Laporte for MTM1) and spectacular results in dog spontaneous model for MTM, A Buj Bello Genethon • A clinical trial on MTM1 babies by Audentes/Genethon in US, 12 children treated with spectacular results (unpublished), and two start up for Friedreich and alternate strategy for MTM1, prepare clinical trials
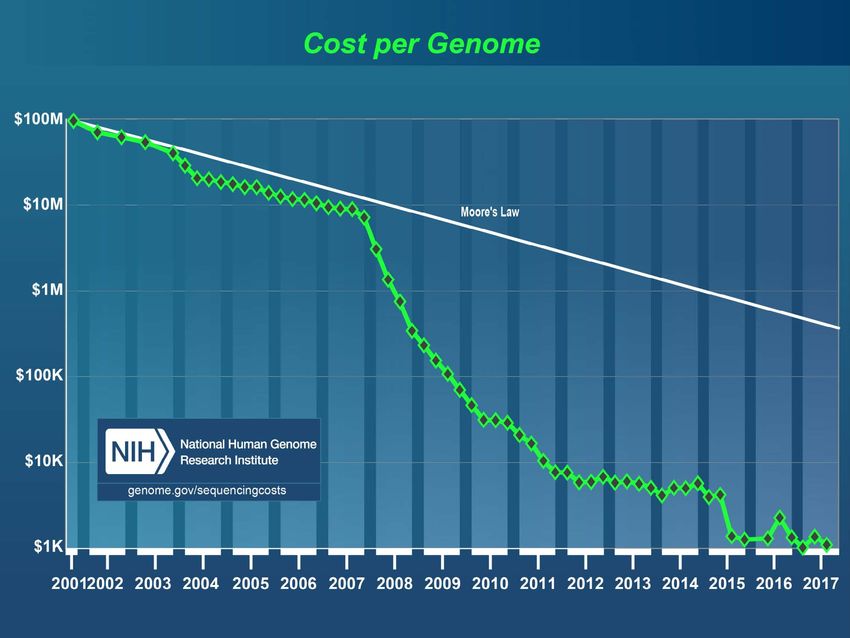
Cette révolution technologique ouvre la voie à des applications de dépistage
génétique préventif en population générale
Loi de Moore
+ Coût du stockage
Séquençage Sanger: un informatique et
amplicon (segment
de l’interprétation
obtenu par PCR) par
réaction, jusqu’à 384 par Séquençage
« run » massivement
parallèle ou
NGSeq
ExomeIntérêt de la détection des mutations pour les maladies génétiques: patients • Diagnostic: savoir, prévoir pour le patient, peut permettre une meilleure prise en charge médicale (dans certains cas thérapeutique spécifique, ex Glut1, CHRN4), éducative, sociale… • Conseil génétique: prévoir, prévenir pour la famille • Prédiction de sévérité souvent limitée: maladies dominantes, mutations faux sens (perte de fonction partielle), gènes modificateurs difficiles à identifier en général • Séquençage à très haut débit permet de développer plus systématiquement le diagnostic des maladies génétiques, y compris pour celles qui sont très hétérogènes (surdité congénitale, rétinite pigmentaire, retard mental monogénique…)
Vous pouvez aussi lire

















































