Imagerie de neurones en profondeur par fibre optique avec champ de vue variable et imagerie à grand champ volumétrique rapide avec sectionnement ...
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Imagerie de neurones en profondeur par fibre optique
avec champ de vue variable et imagerie à grand champ
volumétrique rapide avec sectionnement optique HiLo
Mémoire
François Côté
Maîtrise en biophotonique - avec mémoire
Maître ès sciences (M. Sc.)
Québec, Canada
© François Côté, 2020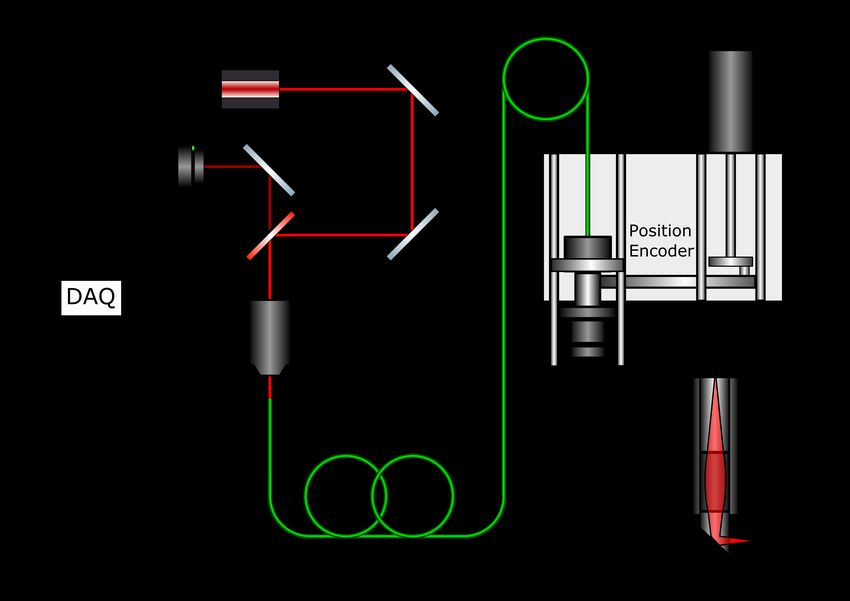
Imagerie de neurones en profondeur par fibre optique
avec champ de vue variable et imagerie à grand
champ volumétrique rapide avec sectionnement
optique HiLo
Mémoire
François Côté
Sous la direction de:
Daniel Côté, directeur de recherche
Martin Lévesque, codirecteur de recherche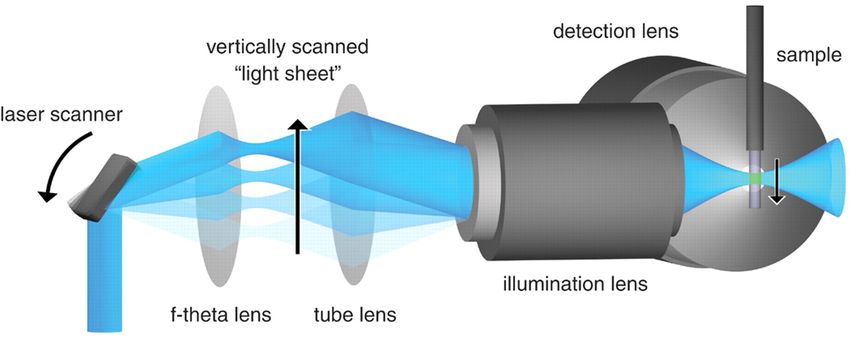
Résumé
Un des défis majeurs en neurosciences est d’acquérir des images de neurones profondément
dans le cerveau tout en gardant un champ de vue raisonnable et en causant le moins de
dommage possible au tissu. Le premier projet décrit dans ce mémoire consiste en un système
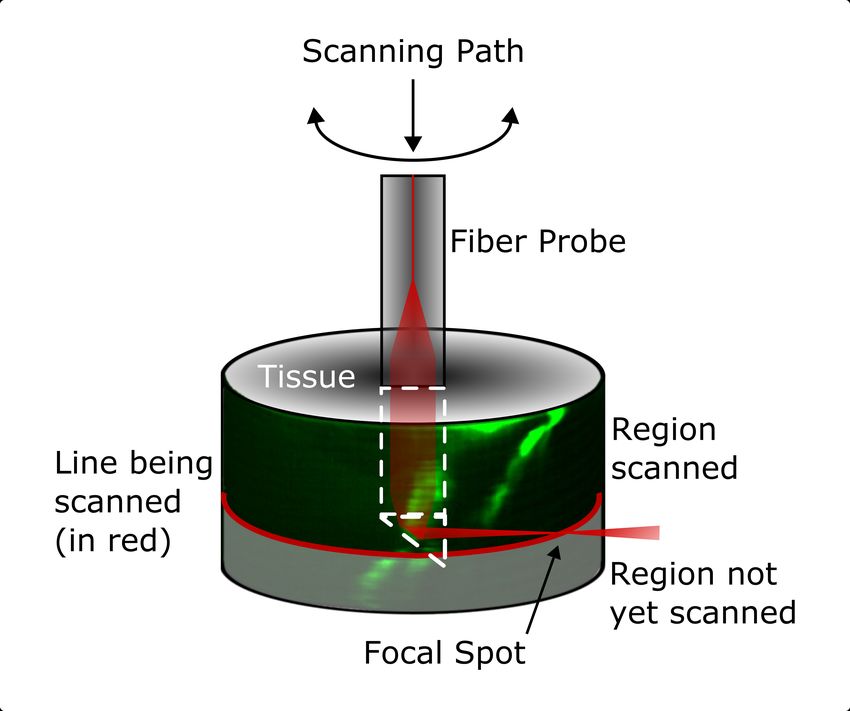
endoscopique à balayage laser. En utilisant des composantes micro-optique au bout d’une
fibre monomode de 125 µm de diamètre, un laser vient être focalisé sur le côté de cette fibre
à environ 60 µm de celle-ci. Un micro capillaire de verre de 250 µm de diamètre externe et
150 µm de diamètre interne sert de guide pour cette fibre dans l’échantillon. Le point focal
possède un diamètre entre 2 et 3 µm et peut être balayé sur une même ligne horizontale
à une vitesse de 30 Hz et sur un angle de 90o par un système construit spécialement pour
suivre la géométrie cylindrique de l’endoscope. Un actuateur piezoélectrique déplace la fibre
verticalement avec une résolution microscopique sur un intervalle de 400 µm pour compléter
une image cylindrique. Le fait de collecter le signal de fluorescence sur le côté de la fibre facilite
son utilisation lors de l’acquisition et permet d’acquérir des images sur des zones non affectées
par la chirurgie. De plus, le champ de vue du système est contrôlé par l’angle de balayage et
la translation verticale de la fibre, donc complètement indépendant du diamètre de celle-ci.
En utilisant des longueurs de fibres à gradient d’indice différentes, il est également possible
de modifier le champ de vue du système, sans modifier son diamètre. Il a été possible avec ce
système d’acquérir des images de microglies dans le mésencéphale d’une souris CX3CR1-GFP.
Le système est prêt à être utilisé pour de l’imagerie calcique sur une ligne de pixel.
L’imagerie de cerveaux entiers peut révéler une panoplie d’informations permettant de mieux
comprendre le développement neuronal à l’échelle microscopique, mais également macrosco-
pique. De plus, visualiser le cerveau comme un tout permet de mieux conceptualiser comment
différentes maladies l’affectent dans son ensemble plutôt que de s’intéresser qu’à certaines
structures spécifiques. En ce moment, il existe deux défis quant à l’imagerie de cerveau de
souris complet : 1) le temps d’acquisition souvent très long (plusieurs heures) et 2) la créa-
tion d’une grande quantité de données qui requiert une gestion ordonnée et spécifique pour ce
genre d’application. Pour pallier à ces défis, on présente dans ce mémoire un système d’ima-
gerie volumétrique à excitation 1-photon ayant une résolution raisonnable, capable d’acquérir
un cerveau de souris entier avec sectionnement optique en quelques minutes. Il s’agit d’une
combinaison entre un système à grand champ et l’algorithme HiLo. Le champ de vue est d’en-
iii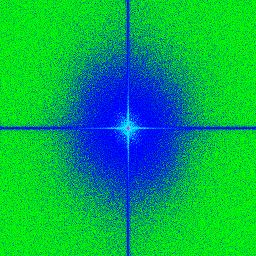
viron 24 mm2 et permet de voir un cerveau de souris âgé d’un jour en presque une image
avec une résolution latérale de 2 µm et une résolution axiale d’environ 4 µm. Cette résolution
permet l’acquisition de corps cellulaires et de projections axonales. Ce système permet donc
d’enregistrer les images dans un format plus facile à utiliser et prenant moins d’espace disque.
Cet outil permettrait de valider la qualité des échantillons volumétriques avant de passer aux
systèmes hautes résolutions. Finalement, puisque ce microscope est rapide, possède un champ
de vue très large et ne requiert pas d’objectifs haute résolution pour l’imagerie 2-photons,
il serait un outil idéal pour des échantillons en "Expansion Microscopy" (technique permet-
tant de grossir les échantillons sans perdre l’intégrité structurelle), technique maintenant très
populaire en neurobiologie.
iv
Abstract
Imaging cells and axons in deep brain with minimal damage while keeping a sizable field of
view remains a challenge, because it is difficult to optimize one without sacrificing the other.
We propose a scanning method reminiscent of laser scanning microscopy to get a reasonable
field of view with minimal damage deep in the brain. By using micro-optics at the tip of our
125 µm-diameter singlemode fiber inside a 250 µm capillary, we can create a focal spot on the
side of the fiber at a distance of approximately 60 µm. The focal spot has a 2 µm diameter
and can be scanned at up to 30 hertz by a custom scanning device over a 90 degree angular
sweep on a single line. A piezoelectric actuator moves up and down the fiber to achieve a
cylindrical scanning pattern. By having this side illumination, there is no need for surgical
exposure of the tissue, making our method simple and easy to achieve. The field of view is
controlled by the angular and vertical sweeps, unrelated to the fiber diameter. Furthermore,
by modifying the length of the grin lens, we could directly increase or decrease the field of
view of our optical system, without any change on the probe diameter. We have succeeded
in imaging microglia in the midbrain of a CX3CR1-GFP mouse. The system is also ready for
calcium imaging on single pixel lines.
Imaging whole mouse brains can provide a wealth of information for understanding neuronal
development at both the microscopic and macroscopic scale. Furthermore, visualizing entire
brain samples allow us to better conceptualize how different diseases affect the brain as a
whole, rather than only investigating a certain structure. Currently, two main challenges exist
in achieving whole mouse brain imaging: 1) Long image acquisition sessions (on the order
of several hours) and 2) Big data creation and management due to the large, high-resolution
image volumes created. To overcome these challenges, we present a fast 1-photon system with
a slightly decreased resolution allowing whole brain, optically sectioned imaging on the order
of minutes by using a mathematical algorithm termed “HiLo”. Our large field of view (25 mm2 )
allows us to see an entire newborn mouse brain in a single snapshot with a resolution of about
2 µm in lateral direction and 4 µm in axial direction. This resolution still allows visualization
of cells and some large axonal projections. This technological advancement will first and
foremost allow us to rapidly image large volume samples and store them in a smaller format
without losing the integral information, which is mainly stained-cell quantity and location.
Secondly, the design will allow for increased successful high-resolution imaging by screening
v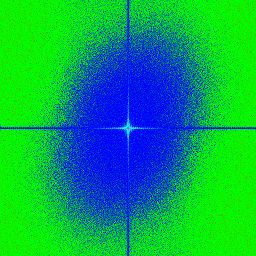
for well-stained samples before commencing their multi-hour acquisitions. Lastly, since the
microscope has a large field of view and does not require collection objectives needed in 2-
photon high resolution microscopes, we can use this device to image expansion microscopy (a
technique which can grow samples while keeping their structural integrity) samples as they
are quickly gaining interest in the biological field.
vi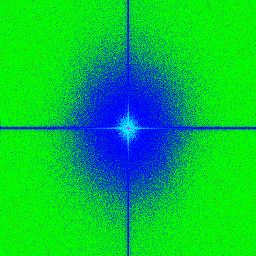
Table des matières
Résumé iii
Abstract v
Table des matières vii
Liste des tableaux ix
Liste des figures x
Remerciements xv
Avant-propos xvii
Introduction 1
1 Imagerie par fluorescence en biologie 6
1.1 Principe de fluorescence . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
1.2 Diffusion de la lumière . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
1.3 Absorption de la lumière . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.4 Faisceau gaussien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.5 Invariant optique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
1.6 Microscopie à grand champ de vue . . . . . . . . . . . . . . . . . . . . . . . 9
1.7 Microscopie à balayage laser . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
1.8 Microscopie par feuillet de lumière . . . . . . . . . . . . . . . . . . . . . . . 11
1.9 Transparisation des tissus . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
1.9.1 iDisco . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
1.9.2 Expansion Microscopy . . . . . . . . . . . . . . . . . . . . . . . . . . 14
1.9.3 SHIELD . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
2 Imagerie longitudinale de neurones in vivo en profondeur 16
2.1 Imagerie par fibre optique . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
2.2 Montage optique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
2.3 Simulations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.4 Logiciel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.5 Caractérisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.5.1 Résolution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 25
2.5.2 Profondeur de champ . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.6 Imagerie dans des tranches de cerveaux ex vivo . . . . . . . . . . . . . . . . 27
vii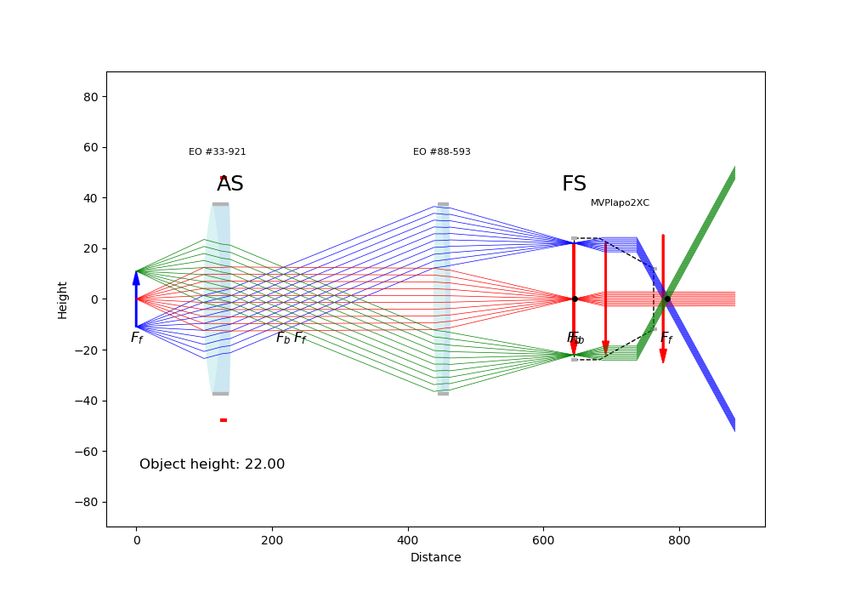
2.7 Imagerie calcique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.8 Imagerie in vivo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
2.9 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
3 Imagerie volumétrique rapide avec sectionnement optique HiLo 37
3.1 Objectifs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
3.2 Les tavelures dans une fibre optique et leurs propriétés . . . . . . . . . . . . 38
3.3 Simulations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.4 Montage optique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
3.5 Développement mathématique du HiLo . . . . . . . . . . . . . . . . . . . . . 43
3.6 Caractérisation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
3.6.1 Taille des grains laser . . . . . . . . . . . . . . . . . . . . . . . . . . 44
3.6.2 Résolution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
3.7 Efficacité de transition du scrambler de GigaConcept . . . . . . . . . . . . . 48
3.7.1 Résumé . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.7.2 Méthode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.7.3 Résultats . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
3.8 Imagerie de cerveaux transparents . . . . . . . . . . . . . . . . . . . . . . . 49
3.9 Discussion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
Conclusion 62
Bibliographie 64
A Codes de simulations du PIM 70
A.1 Sans capillaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
A.2 Avec capillaire . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
B Codes de simulations du HiLo 72
C Étude sur l’efficacité de transition du scrambler de GigaConcept 73
C.1 Fréquence d’allumage on/off du scrambler de 12.5Hz . . . . . . . . . . . . . 73
C.2 Fréquence d’allumage on/off du scrambler de 25Hz . . . . . . . . . . . . . . 73
C.3 Codes - Mesure du temps de réponse du scrambler . . . . . . . . . . . . . . 73
C.4 Codes - Mesure de la fréquence maximal d’utilisation du scrambler . . . . . 75
C.5 Codes - Tests reliés à l’étude du scrambler . . . . . . . . . . . . . . . . . . . 77
viii
Liste des tableaux
3.1 Durée de la période transitoire du scrambler pour passer de l’étant uniforme à
des tavelures pour différentes acquisitions. . . . . . . . . . . . . . . . . . . . . . 49
3.2 Taux de réussite du scrambler à passer d’un état uniforme à des tavelures à diffé-
rente fréquence d’oscillation on/off. On remarque que typiquement, la fréquence
maximale d’utilisation est de 15 Hz, soit 7,5 images HiLo par seconde. . . . . . 50
ix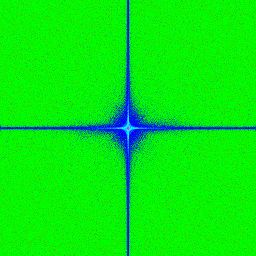
Liste des figures
1.1 Diagramme de Jablonski démontrant le concept de fluorescence.[4] . . . . . . . 7
1.2 Schéma d’un faisceau gaussien présentant ces caractéristiques.[5] . . . . . . . . 9
1.3 Le produit de la surface de la source et son cône forme un invariant optique.[38] 10
1.4 Microscopie de fluorescence à champ large.[6] . . . . . . . . . . . . . . . . . . . 11
1.5 Principe de la microscopie confocal.[3] . . . . . . . . . . . . . . . . . . . . . . . 12
1.6 Feuillet de lumière créé à partir de lentilles cylindriques.[12] . . . . . . . . . . . 13
1.7 Feuillet de lumière créé par balayage d’un faisceau gaussien.[23] . . . . . . . . . 13
2.1 Comparaison entre 3 types de fibre optique pour l’imagerie et la microscopie. . 17
2.2 De la micro-optique est ajoutée au bout de la fibre pour permettre son utilisation
dans un système à balayage imageant. . . . . . . . . . . . . . . . . . . . . . . . 19
2.3 Schéma du montage optique du PIM . . . . . . . . . . . . . . . . . . . . . . . . 19
2.4 Représentation schématique de la reconstruction de l’image . . . . . . . . . . . 21
2.5 Le champ de vue du système n’est plus dépendant du diamètre de la fibre, mais
de son système de balayage et de sa distance focale. . . . . . . . . . . . . . . . 22
2.6 Tracer de rayons depuis la sortie de la fibre optique jusqu’au point focal, passant
par les 2 barreaux de verre et la GRIN. . . . . . . . . . . . . . . . . . . . . . . 22
2.7 Tracer de rayons depuis la sortie de la fibre optique jusqu’au point focal, passant
par les 2 barreaux de verre et la GRIN en considérant un capillaire de verre de
150 micromètres de diamètre interne et 250 micromètres de diamètre externe. . 23
2.8 Diagramme fonctionnel du logiciel d’acquisition du microscope. . . . . . . . . . 24
2.9 Microbilles fluorescentes de 3-4 micromètres en diamètre. Image acquise par le
microscope à fibre PIM. La barre correspond à 30 micromètres. . . . . . . . . . 25
2.10 Profile d’intensité de 10 billes fluorescentes acquises avec un microscope confocal
Zeiss LSM 710 et avec le PIM. . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.11 Amplitude normalisée en fonction de la distance séparant la fibre optique de la
lame fluorescente. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
2.12 Image prise dans la substance noire d’un cerveau de souris adulte marqué Anti-
TH avec le fluorophore Alexa594. La barre d’échelle correspond à 60 micromètres. 28
2.13 Profil d’intensité d’un filament dendritique et d’un soma d’un neurone dopami-
nergique dans la substance noire d’un cerveau de souris adulte avec un marquage
anti-TH. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
2.14 Signaux spontanés d’une tranche organotypique d’hippocampe de rat ayant été
marquée avec un virus GCAMP6s. . . . . . . . . . . . . . . . . . . . . . . . . . 30
2.15 Un microcapillaire de verre est utilisé comme guide pour l’imagerie in vivo. . . 31
2.16 Microglies dans le mésencéphale d’une souris CX3-CR1-GFP. . . . . . . . . . . 32
2.17 Microglies dans le mésencéphale d’une souris CX3-CR1-GFP. . . . . . . . . . . 33
x2.18 Microglies dans le mésencéphale d’une souris CX3CR1-GFP. . . . . . . . . . . . 34
3.1 L’algorithme HiLo utilise deux types d’illumination laser pour fonctionner. . . . 38
3.2 Deux modes se propageant dans une fibre optique vont produire un patron
d’interférence. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
3.3 Simulation par tracé de rayons de l’illumination du microscope HiLo. . . . . . . 41
3.4 Montage optique du microscope HiLo à grand champ de vue. . . . . . . . . . . 43
3.5 Les grains sont visibles après avoir appliqué un filtre passe-bas sur l’image prise
avec une lame de microscope fluorescente. . . . . . . . . . . . . . . . . . . . . . 45
3.6 Les grains laser ne sont pas visibles sur l’image avec une illumination uniforme,
comme il est souhaité, même après avoir passé un filtre passe-bas. . . . . . . . . 46
3.7 a,b,c,d) Transformée de Fourier rapide ("Fast Fourier Transoform") d’images
avec une illumination par tavelures provenant de différentes fibres avec des ou-
vertures numériques (NA) variables. Le centre correspond aux basses fréquences
et les extrémités de l’image correspondent aux hautes fréquences. . . . . . . . . 54
3.8 Une cible Thorlabs R1DS1N est utilisée pour caractériser la résolution du système. 55
3.9 Les éléments 3 du groupe 7 sont les plus petits éléments que le microscope peut
résoudre. Les éléments du groupe 4 deviennent trop flous. On voit que l’intensité
lumineuse de chaque ligne n’est pas constante. . . . . . . . . . . . . . . . . . . 55
3.10 Trois images HiLo consécutives d’une bille fluorescente de 3-4 µm de diamètre.
Chaque image est séparée de 5 µm dans l’axe axial. Les figures d, e et f sont le
profil d’intensité correspondant pour chaque image sur la ligne présentée en a. . 56
3.11 Variance d’une série de données en fonction du temps. Vers 1 seconde, le scram-
bler est allumé et la variance est donc élevée. À 2 secondes, le scrambler est
arrêté et la variance passe à zéro. . . . . . . . . . . . . . . . . . . . . . . . . . . 56
3.12 Données de variance à partir du dernier pic de la figure 3.11 avec un fit. Le
temps caractéristique correspond au temps pour descendre à 10% de la valeur
de variance maximale. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
3.13 Cerveau de souris P1 avec marquage anti-TH Alexa594 avec deux types d’illu-
mination. La barre correspond à 1 mm. . . . . . . . . . . . . . . . . . . . . . . 57
3.14 Image HiLo d’un cerveau de souris P1 avec un marquage anti-TH et le fluoro-
phore Alexa594. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
3.15 Zoom sur des filaments axonaux avec l’illumination uniforme et HiLo, ainsi que
leur profile d’intensité correspondante. La barre équivaut à 500 micromètres. . . 59
3.16 Cerveau de souris P1 avec marquage anti-TH Alexa594 avec les deux types
d’illumination. La barre équivaut à 1 millimètre. . . . . . . . . . . . . . . . . . 59
3.17 Image HiLo d’un cerveau de souris P1 avec un marquage anti-TH et le fluoro-
phone Alexa594. La barre équivaut à 1 millimètre. . . . . . . . . . . . . . . . . 60
3.18 Zoom sur une cellule dopaminergique située dans la région hypothalamique
antérieure avec son profil d’intensité. . . . . . . . . . . . . . . . . . . . . . . . . 60
3.19 Zoom sur une cellule dopaminergique située dans la substance noire avec son
profil d’intensité. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61
3.20 Vue d’un mésencéphale sur un hémisphère de cerveau de souris adulte en coupe
sagittal avec un marquage SHIELD anti-TH. . . . . . . . . . . . . . . . . . . . 61
C.1 Données de variance lorsque le scrambler est allumé et éteint à une fréquence
de 12.5Hz. On peut voir que les données descendent à zéro de variance, donc à
un état speckle. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
xiC.2 Données de variance lorsque le scrambler est allumé et éteint à une fréquence
de 25Hz. On peut voir que les données ne descendent pas à zéro de variance,
donc à un état speckle complet. . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
xiixiii
There is no unique picture of
reality
Stephen Hawking
xivRemerciements
Je souhaite tout d’abord adresser toute ma gratitude à mon directeur de recherche Daniel
Côté pour le temps qu’il m’a consacré malgré son horaire chargé. Mon séjour dans son labora-
toire m’a beaucoup apporté, tant sur le plan scientifique que personnel. Ses nombreux conseils
ont su nourrir mes réflexions et à pousser plus loin mes recherches. Il a su me guider dans
cette grande aventure avec passion et rigueur. C’est avec cette même passion que je souhaite
puiser mon inspiration dans le futur.
Je désire remercier mon codirecteur Martin Lévesque pour son écoute et sa disponibilité. Il
m’a inclus dans son laboratoire même si je n’avais aucune connaissance en neurobiologie. Tou-
tefois, je m’y suis toujours senti le bienvenu et m’a permis de mieux appréhender l’importante
cohésion entre l’ingénierie et la science fondamentale. Les échantillons biologiques proviennent
tous de son laboratoire et je n’aurais pas pu compléter ces deux projets sans son aide et celle
de son équipe.
I would like to thank Damon DePaoli, my laboratory partner. This journey would not have
been the same without him. His infinite curiosity and his intuition has always impressed me.
His feedback on my results have been good checkpoints during the project development and
were super helpful. Thanks you so much !
Un merci spécial à Véronique Rioux et Anne Sebilo pour leur aide avec les chirurgies des
souris et la préparation des expériences in vivo. Travailler avec des prototypes de microscope est
une chose, mais y impliquer des animaux vivants en est une autre. Leur aide fut fondamentale
et très appréciée.
J’aimerais remercier les différents stagiaires du laboratoire à Daniel Côté pour l’énorme
travail qu’ils ont fait pendant leur court passage. Ils ont fait progresser beaucoup de projets
en suspens et plusieurs ont pu être finalisés. Je voudrais personnellement remercier Ludovick
Bégin qui a programmé le logiciel d’acquisition du PIM (Aleph) en Python à partir de la
librairie National Instruments. Il a relevé le défi à merveille et son aide fut primordiale pour
le bon fonctionnement du projet.
xvJe voudrais exprimer ma reconnaissance envers les amis et collègues qui m’ont apporté leur
support moral et intellectuel tout au long de ma démarche. Un grand merci à Charles Gora
pour toutes ses heures passées ensemble au microscope HiLo et au lightsheet.
Enfin, merci à mes parents pour leur soutien inconditionnel et à mes trois soeurs pour leur
encouragement.
xviAvant-propos
Ce mémoire est issu de ma maîtrise en biophotonique que j’ai réalisée dans les laboratoires
des professeurs Daniel Côté et Martin Lévesque au centre de recherche CERVO à Québec. Mon
projet portait sur le microscope à fibre PIM (« Panoramic Interstitial Microscope ») présenté
au chapitre 2. Ce projet avait déjà été commencé par Joël Crépeau lors de sa maîtrise et a par
la suite été repris par Nicolas Lapointe. Le but de mon projet était alors de rendre le système
fonctionnel au laboratoire, de l’optimiser pour prendre des images in vivo dans des souris
vivantes et de développer une méthode pour enregistrer l’activité calcique. Cette expérience
m’a fait comprendre beaucoup de notions sur l’imagerie in vivo et sur l’ingénierie de système.
Un article scientifique devrait sortir par rapport à ces résultats.
En commençant ma maîtrise, j’ai tout de suite été impliqué dans un second projet d’imagerie
volumétrique avec sectionnement optique HiLo en utilisant un module à fibre optique. Le
projet a bien fonctionné et nous avons pris la décision d’ajouter ce projet à mon mémoire.
Le microscope a donc été optimisé pour l’imagerie volumétrique de cerveaux de souris. La
transparisation des échantillons est nécessaire pour ces travaux, ce qui m’a apporté beaucoup
de connaissances dans ce domaine d’étude. Ce projet devrait également mener à un article
scientifique dans un futur proche.
Mon expérience sur la manipulation d’échantillons volumétriques transparents m’a permis
d’être approché pour devenir responsable du microscope à feuillet de lumière du centre de
recherche CERVO. Ce microscope a été développé par Cléophace Akitegetse pendant son doc-
torat avec le professeur Daniel Côté en collaboration avec le professeur Martin Lévesque. Le
microscope avait été utilisé principalement avec des échantillons idisco. Suite au départ de
Cléophace à la fin de son projet, j’ai donc pris le relais de l’imagerie volumétrique avec ce
microscope afin d’optimiser son utilisation avec d’autres méthodes de transparisation, comme
clarity, SHIELD et « Expansion Microscope ». J’ai pris également soin de montrer son fonction-
nement à quelques biologistes du groupe à Martin Lévesque pour que le microscope continue
d’être utilisé suite à mon départ. Les résultats de ces expériences ne sont pas présents dans ce
mémoire, mais feront l’objet d’une publication.
xviiCe passage au centre de recherche CERVO était une expérience enrichissante en termes de
connaissances générales sur la microscopie et sur la recherche et développement. J’ai pu me
forger une méthode de travail et développer des aptitudes en gestion de projet. La microscopie
est un domaine fascinant qui oblige une certaine cohésion entre l’ingénierie et la science fonda-
mentale. C’est un pont qui nous permet de jeter un regard direct sur les applications possibles
des technologies que nous développons, ce qui nous encourage toujours à faire mieux.
xviiiIntroduction
La microscopie a beaucoup évolué depuis les dernières décennies et ne cesse de susciter
l’intérêt des chercheurs, tant dans les sciences de la vie que dans les sciences physiques. C’est un
outil puissant qui permet de répondre à plusieurs questions, et ce, grâce à la lumière. L’optique
est un domaine en constante expansion depuis la deuxième moitié du 20e siècle avec l’arrivée
du laser. C’est grâce à la lumière et à ses propriétés que l’on doit la naissance de plusieurs
outils telle que la microscopie par fluorescence, indispensable aujourd’hui en neurobiologie.
En effet, la lumière peut être considérée à la fois comme une onde ou une particule que l’on
appelle un photon. L’aspect ondulatoire est directement relié au concept de résolution à la
microscopie par fluorescence avec le critère de Rayleigh, tandis que l’aspect particulaire aide
à comprendre la détection avec les tubes photomultiplicateurs utilisés en microscopie confocal
ou multi photon. Heureusement, les structures biologiques cellulaires et intracellulaires ont une
grandeur de l’ordre du micron, soit environ le même ordre de grandeur que la lumière visible
et proche infrarouge où la recherche dans le domaine a permis la création de plusieurs sources
laser dans cette région du spectre. Il est ainsi possible d’élaborer des systèmes optiques à la
fine pointe de la technologie utilisant ces sources pour résoudre des structures microscopiques,
voir nanoscopique.
La synthèse et l’optimisation de la protéine GFP ("Green Fluorescent Protein") [18] ont fait
naître un domaine d’étude qui à ce jour est vaste et toujours en expansion. La microscopie par
fluorescence a littéralement révolutionné les façons de procéder dans les laboratoires recherches
et la conception de nouveaux appareils est toujours une nécessité. Depuis sa création en 1994
par Roger Tsien, plusieurs dérivés ont fait leur apparition permettant l’utilisation de diverses
longueurs d’onde d’excitation à travers le spectre visible. Il existe désormais une panoplie de
ces marqueurs fluorescents sur le marché et il en existe même dont l’émission dépend de la
concentration de calcium, comme le GCAMP6s et GCAMP6f [34]. Ce dernier est fort utile
pour l’imagerie in vivo.
En neurobiologie, les protéines fluorescentes servent à tout moment dans de multiples appli-
cations. Leur fréquente utilisation force parfois les chercheurs a créer des outils plus spécifiques
à leurs besoins pour répondre aux questions qu’ils se posent. C’est le cas pour l’imagerie in vivo
1ou l’imagerie profonde dans le cerveau où les outils étaient très limités par le passé. Encore
aujourd’hui, beaucoup de défis restent à surmonter en ce qui concerne ce genre de technique,
en raison du risque de la procédure et de la diffusion de la lumière dans le tissu augmentant
la perte du signal d’excitation et de collection.
Un outil ayant révolutionné la recherche en neurosciences est sans doute la microscopie mul-
tiphotonique [19, 54]. Il est possible notamment avec cette technique d’observer et d’analyser
l’activité neuronale dans un animal vivant. La microscopie par absorption 2-photon permet
de prendre des images à plusieurs centaines de micromètres sous la surface du cortex sans
être gêné par la diffusion de la lumière. L’arrivée de la microscopie 3-photons a fait grimper
cette limite à près de 1,5 mm sans endommager le tissu. De plus, l’absorption multiphoto-
nique procure un sectionnement optique de par sa nature non linéaire et sa faible probabilité
de se manifester dans l’échantillon. Ce genre de microscopie permet notamment d’obtenir des
résolutions spatiales de l’ordre d’une fraction de micromètre. De ce fait, la microscopie mul-
tiphotonique a difficilement accès aux structures subcorticales. D’autres alternatives doivent
être employées pour imager dans ces régions du cerveau.
C’est dans ce contexte que des techniques un peu plus invasives ont été inventées. Une de
ces approches implique l’utilisation de lentilles GRIN ("gradient index") pour relayer le plan
focal de l’objectif à un plan conjugué plus en profondeur. Ces lentilles ont la particularité
d’avoir un changement d’indice de réfraction graduel et périodique sur leurs longueurs, ce qui
permet d’obtenir relativement facilement un système de relais dans un barreau de verre. Il
est possible de se procurer des lentilles GRIN relativement petites pour être utilisées dans un
contexte chirurgical. Ainsi, ces lentilles peuvent être implantées dans le cerveau sans trop de
dommage et relayer l’information d’un bout à l’autre de la lentille sur toute sa longueur jusqu’à
un objectif et un système d’acquisition standard [44]. Éventuellement, ce genre de technique
a gagné en popularité et a pu être utilisé pour enregistrer l’activité neuronale [44], effectuer
de la photostimulation dans des souris libres de mouvement [51] ou simplement de l’imagerie
2-photon [2] en profondeur pour ne nommer que quelques exemples. La lentille GRIN a fait
ses preuves en tant que solution viable pour l’imagerie en profondeur dans le cerveau sous
différents modes d’opération. Or, il demeure que l’insertion d’un barreau de verre dans un
cerveau de souris de cette taille (environ 0,5 millimètre pour les plus petits en diamètre) est
dommageable et peut être problématique lors de la chirurgie. De plus, les performances de
ce système sont souvent proportionnelles au diamètre de la lentille. Il y a donc forcément
un équilibre à respecter entre les spécifications du système et le dommage qu’il va causer à
l’animal, souvent irréversible.
Une autre méthode fort populaire est l’utilisation de composantes à fibre optique. Le princi-
pal avantage des fibres optiques est leur petit diamètre. Ce dernier critère rend leur utilisation
pour l’imagerie in vivo beaucoup plus permissive et facile à implémenter. La plupart uti-
2lisent des fibres optiques multimodes ou des fibres multicoeurs et interprètent les résultats
comme un système à grand champ de vue [39, 55]. Ces systèmes peuvent permettre une ré-
solution allant jusqu’au micromètre avec évidemment un champ de vue proportionnel à leur
diamètre. Ces fibres peuvent également permettre une analyse en photométrie de l’activité
neuronale [17]. Toutefois, les fibres multimodes à un coeur peuvent être très difficiles à utiliser
dans un contexte in vivo, puisque le phénomène d’interférence contenu dans le coeur doit être
parfaitement maîtrisé et connu pour éventuellement reconstruire une image. Il est également
possible d’enregistrer l’activité neuronale en photométrie avec des matrices de fibres.
Il est possible de combiner ces deux outils, les fibres optiques multimodes et les lentilles
GRIN, pour obtenir des systèmes aussi performants [14, 16]. Ce type d’endoscope est fortement
utilisé en tomographie par cohérence optique [52, 29, 30].
Tous ces systèmes utilisent des fibres multimodes, mais les fibres monomodes sont très
rarement utilisées. En effet, si on revient à un système à grand champ, une fibre monomode
serait l’équivalent de l’enregistrement d’un pixel dans une image. Il serait possible d’utiliser ce
système pour de la photométrie, mais l’efficacité de collection est trop faible comparativement
à une fibre multimode beaucoup plus facile à appliquer dans ce genre d’application. Toutefois,
en combinant la géométrie de la fibre optique avec un système approprié, il serait possible
d’obtenir un endoscope semblable à un microscope confocal. Le fait que le laser sort sur le
côté de la fibre optique rend le système très versatile et offre beaucoup d’avantages, notamment
sur le champ de vue variable du système. Le système a été appelé le "Panoramic Interstitial
Microscope" ou PIM et serait très prometteur pour l’imagerie profonde de neurones dans le
cerveau.
Même si les expériences dans les animaux vivants peuvent procurer au chercheur une grande
quantité d’information pertinente, toute la gestion autour de cette procédure est coûteuse et
peut souvent être compliquée à se concrétiser. Souvent, il peut être beaucoup plus avantageux
de travailler sur des échantillons ex vivo. C’est le cas lorsqu’on s’intéresse à des structures
beaucoup plus volumineuses qui s’étendent sur plusieurs régions du cerveau, comme le système
dopaminergique. En effet, acquérir un cerveau de souris complet procure une grande quantité
d’information permettant de mieux comprendre le développement neurologique à l’échelle
microscopique, mais également macroscopique. De plus, une image volumétrique d’un cerveau
entier permet aux chercheurs de mieux conceptualiser les liens de causes à effets de différentes
maladies qui affectent le cerveau comme un tout, plutôt que de s’interroger seulement des
conséquences sur quelques régions.
La méthode la plus populaire à ce jour pour procéder à l’imagerie d’échantillons volumé-
trique est sans doute la microscopie par feuillet de lumière. Elle a été utilisée en neurosciences
pour la première fois en 2004 pour l’imagerie d’embryons [20] et n’a cessé de gagner l’intérêt
3des groupes de recherche partout à travers le monde, de par sa simplicité d’utilisation et de la
qualité des images qu’il est possible d’enregistrer. C’est un outil puissant permettant la visua-
lisation du réseau de neurones dans un cerveau de souris entier [12]. Depuis, cette technique a
fait place à plusieurs variantes comme le feuillet à balayage [23], une utilisation en excitation
2-photons [48], puis en 3-photons [13]. Un des défis majeurs de ce cette technique est la ré-
solution axiale souvent moins performante que la résolution latérale. En raison des propriétés
des faisceaux gaussiens, l’épaisseur du feuillet ne sera jamais uniforme. La résolution latérale
peut atteindre des valeurs de quelques centaines de nanomètres, tandis que la résolution axiale
varie normalement entre 4 et 20 µm [28]. Un compromis est inévitable, puisque la longueur de
la feuille sera proportionnelle à l’inverse de la résolution axiale. Il est possible de remédier à
cette situation en utilisant un faisceau de bessel à l’excitation plutôt que faisceau gaussien. Ce
type de faisceau permet entre autres de former une longue ligne dont l’épaisseur est uniforme
et peut atteindre un diamètre de l’ordre du micromètre. En utilisant cette ligne avec un sys-
tème à balayage, il est possible d’obtenir un feuillet de lumière capable de fournir au système
d’imagerie une résolution isotropique.
Un cerveau de souris ayant un volume d’environ 250 mm3 peut générer près de 1 To de don-
nées et demande plusieurs heures d’acquisition. Ce genre d’imagerie requiert donc un système
de gestion et de traitement des données complet et robuste. Le temps entre la préparation
de l’échantillon et l’analyse des images peut prendre plusieurs semaines. Les délais d’attente
peuvent augmenter dans le cas où le marquage de l’échantillon n’a pas fonctionné ou si l’échan-
tillon n’était pas assez de bonne qualité. Ce genre de situation peut devenir assez récurrent
lorsque les chercheurs essayent de nouveaux protocoles. Ainsi, pour prévoir la qualité d’un
échantillon, il peut être très pratique d’utiliser un système à grand champ de vue. C’est une
méthode qui est rapide qui demande très peu de temps d’acquisition et qui génère très peu
de données comparativement aux systèmes par feuillets de lumière. Cependant, ce genre de
microscopie est très peu utilisé pour des échantillons volumétriques en raison de l’absence de
sectionnement optique sur ces systèmes. Les images sont donc pour la plupart floues, puisque
la lumière provenant de tout l’échantillon est enregistrée simultanément. Or, ce problème peut
se résoudre grâce à la technique HiLo [56, 27]. En combinant ces deux méthodes d’imagerie,
on obtient un microscope capable d’acquérir un cerveau de souris entier rapidement de par
son grand champ de vue de près de 35 mm2 . Cet instrument serait un outil de choix pouvant
prédire la qualité des échantillons volumétriques rapidement pour l’imagerie haute résolution.
La microscopie par feuillet de lumière et la microscopie à grand champ de vue de volumes
requièrent toutes deux que l’échantillon soit le plus transparent possible, sans quoi le laser ne
pourrait pas pénétrer le tissu et exciter les protéines fluorescentes dans toutes les structures
du cerveau. L’invention des méthodes de transparisation a permis d’améliorer grandement
la qualité des images et d’améliorer la recherche en neurosciences en général. Il existe plu-
sieurs protocoles ayant chacun leurs avantages et leurs inconvénients, les plus populaires étant
4iDisco [45], clarity [10] et plus récemment l’« Expansion Microscopy » [7], ayant un intérêt
grandissant en neurosciences. Cette dernière méthode fonctionne sur le principe de super-
résolution en grossissant l’échantillon plutôt que de repousser les limites de résolution de la
microscopie par fluorescence. Pour donner une idée de comparaison, un cerveau de souris
adulte (ayant un volume d’environ 1,5 cm3 ) peut grossir jusqu’à un volume semblable à une
balle de golf. Ainsi, toutes les structures qui composent le tissu peuvent grossir jusqu’à dix fois
leur taille originale, ce qui revient à avoir une résolution dix fois meilleure. À ce jour, l’imagerie
de tels échantillons s’effectue sur des tranches seulement, en raison du temps d’acquisition et
la génération de donnée très élevés. La distance de travail des objectifs haute résolution en
excitation et en collection des systèmes par feuillets de lumière limite l’imagerie à de petits
volumes seulement, rendant impossible l’acquisition d’échantillons complets en « Expansion
Microscopy ». C’est pour cette raison que l’on pense que le système à grand champ de vue
jumelé à la technologie HiLo sera un candidat idéal pour ce genre d’imagerie.
Pour tester les systèmes dans une application biologique, on s’est intéressé particulièrement
au système dopaminergique. L’impact de ces neurones est étudié en neurosciences sur plu-
sieurs maladies neurodégénératives comme la maladie de Parkinson. En effet, le principal lien
de causalité avec cette maladie provient de la dégénérescence des neurones dopaminergiques
de la substance noire dans le mésencéphale [1]. Ils ont un rôle primordial dans la locomotion,
puisqu’ils sont impliqués dans la voie nigrostriée du système dopaminergique [11]. Leur dégé-
nérescence amène alors une réduction de la dopamine jusque dans les régions du cerveau où
ils projettent, soit le striatum, impliqué dans le contrôle moteur [9]. Les neurones dopaminer-
giques peuvent donc être étudiés sous plusieurs formes. Tout d’abord, on peut examiner ces
neurones localement dans le mésencéphale, ce qui requiert un système d’imagerie assez petit
et versatile pour aller en profondeur dans le cerveau sans causer trop de dommage. Enfin, la
globalité du système peut également être étudiée dans le cerveau, ce qui demande un micro-
scope capable d’acquérir des images en trois dimensions rapidement sur un très grand champ
de vue. Le système dopaminergique est donc un modèle d’étude idéal pour tester les deux
microscopes qui seront discutés dans ce mémoire. Ces nouveaux outils pourront par la suite
être utilisés en recherche fondamentale telle que la recherche sur maladie de Parkinson.
5Chapitre 1
Imagerie par fluorescence en biologie
1.1 Principe de fluorescence
La fluorescence est un principe physique qui peut s’expliquer à partir des niveaux d’énergie
des molécules. Le diagramme de Jablonski est souvent utilisé pour schématiser ce phénomène,
comme on peut le voir à la figure 1.1. De manière générale, la thermodynamique prédit qu’une
molécule va demeurer à l’état stable électroniquement, puisque la différence d’énergie entre
le niveau fondamental et les niveaux excités est beaucoup plus grande que le rayonnement
thermique. Donc, pour passer du niveau fondamental à un niveau excité, les électrons doivent
absorber de l’énergie. Ceci peut se concrétiser par l’absorption d’un ou plusieurs photons
incidents dont l’énergie totale correspond à la différence d’énergie entre le niveau fondamental
et le niveau excité. Les électrons excités vont tenter de revenir à un état stable en passant à des
niveaux d’énergie inférieurs dans la molécule. Lorsque l’électron revient au niveau fondamental,
celui-ci relâche son énergie sous forme de lumière en émettant un photon. Puisque l’énergie
émise est moins élevée que l’énergie qui a été nécessaire pour l’excitation, le photon émis aura
donc une longueur d’onde plus élevée, donc moins énergétique.
En biologie, la fluorescence est la méthode de contraste la plus répandue en recherche. Il est
possible de créer des protéines fluorescentes qui peuvent se fixer sur des structures d’intérêt à
l’aide d’anticorps spécifiques. Elle est surtout utilisée pour la microscopie à fluorescence par
excitation monophotonique et multiphotonique. Dans le premier cas, un photon est absorbé
pour exciter la molécule et un photon de moins grande énergie est relâché. En excitation
multiphotonique, plusieurs photons sont absorbés et un photon est émis par la suite. Toutefois,
la probabilité que plusieurs photons soient absorbés au même moment est très faible, d’où le
fait que ce phénomène se produit seulement au point focal provenant d’un laser avec une
grande irradiance, ce qui confère à cette technique son sectionnement optique. C’est pour
cette raison que des lasers à impulsions sont couramment utilisés pour ce genre d’expérience.
Pour la même molécule, le photon émis a la même énergie, qu’il ait été créé en excitation
6Figure 1.1 – Diagramme de Jablonski démontrant le concept de fluorescence.[4]
monophotonique ou en multiphotonique. De plus, l’énergie totale des photons absorbés lors
de l’excitation doit correspondre à la différence d’énergie entre le niveau excité et le niveau
fondamental.
1.2 Diffusion de la lumière
La diffusion de la lumière est un phénomène physique décrivant le changement de direction
de la propagation de la lumière résultant de son interaction avec la matière. La diffusion peut
se décrire de plusieurs façons. La première étant que la lumière interagit avec des particules
ayant un indice de réfraction différent du milieu de propagation et la deuxième comme étant
une propagation dans un milieu ayant un indice de réfraction continu, mais fluctuant[21].
La diffusion dépend de plusieurs composantes, comme la taille des particules diffusantes, la
longueur d’onde ou la variation de l’indice de réfraction dans le tissu, etc.
Puisque la lumière diffusée va nécessairement changer de direction et ne va pas nécessaire-
ment être captée par le détecteur, il existe une équation décrivant l’intensité non diffusée It
dans un échantillon d’épaisseur d alors qu’il est éclairé par une source d’intensité Io .
Id = Io · e−µs d (1.1)
Le coefficient de diffusion µs représente la probabilité qu’un photon de longueur d’onde λ
7diffuse par unité de longueur. Ainsi, au-delà une certaine épaisseur, toute la lumière sera
diffusée dans l’échantillon.
1.3 Absorption de la lumière
Dans un milieu non diffusant, l’énergie des photons d’un faisceau lumineux est absorbée par
les électrons des atomes qui composent le milieu. Cette énergie est alors transformée pour la
plupart sous forme de chaleur dans milieu. Comme la diffusion, les matériaux absorbants sont
caractérisés par un coefficient d’absorption µa . Il est possible de définir l’intensité lumineuse
transmise It dans un milieu d’épaisseur d avec une intensité initial Io .
Id = Io · e−µa d (1.2)
1.4 Faisceau gaussien
Un faisceau gaussien est un faisceau qui est généré à la sortie d’une source laser. L’amplitude
du champ électrique que possède ce faisceau peut être décrite comme une fonction gaussienne
et possède donc une symétrie radiale très caractéristique. Ce type de faisceau est très bien
connu du milieu scientifique, ce qui est très utile pour la simulation. De plus, les faisceaux
lasers divergent très peu, ce qui signifie qu’il est acceptable d’utiliser l’approximation paraxiale.
Siegman décrit l’amplitude du champ électrique d’un faisceau gaussien selon l’équation 1.3[49]
et le rayon de courbure complexe selon l’équation 1.4.
r2
ω0 − ω(z) r2
2 −jk 2R(z) −jkz+jψ(z)
E(r, z) = E0 e e (1.3)
ω(z)
1 1 λ
≡ −j 2 (1.4)
q̃(z) R(z) πω (z)
où
r
z
ω(z) = ω0 1 + ( )2 (1.5)
zr
2
zR
R(z) = z + (1.6)
z
z
ψ(z) = tan−1 ( ) (1.7)
zR
8Figure 1.2 – Schéma d’un faisceau gaussien présentant ces caractéristiques.[5]
ωo2 π
zR = (1.8)
λ
L’étranglement d’un faisceau gaussien est défini comme étant ωo . L’amplitude du faisceau
gaussien est sensiblement constante sur l’intervalle b, où b = 2 · zR . La position où la largeur
√
du faisceau équivaut à 2 · ωo est zR (figure 1.2).
1.5 Invariant optique
Le concept d’invariant optique est fondamental en conception optique. La surface d’une
source par rapport à l’ouverture numérique de cette même source est un invariant dont il
faut tenir compte dans l’élaboration du système d’illumination. Dans un système optique où
une source produit une image, le produit de la surface de la source et de son cône d’émission
est proportionnel au produit de la surface et le cône d’illumination de l’image (Figure 1.3).
Cette dépendance peut se décrire par l’équation 1.9. Ce concept sera très important lors de la
conception de systèmes d’illumination.
As Ω s = Ai Ω i (1.9)
1.6 Microscopie à grand champ de vue
La microscopie à champ large est une méthode par fluorescence qui tient sa force dans la
rapidité d’exécution. Une source lumineuse (habituelle une lampe au Xénon) est tout d’abord
filtrée pour ne laisser passer que la longueur d’excitation des protéines fluorescentes de l’échan-
tillon. Cette lumière est focalisée grossièrement par l’objectif qui permet habituellement d’illu-
miner uniformément l’échantillon sur un grand champ de vue. Le signal fluorescent est alors
capté par l’objectif et filtré par le miroir dichroïque et un filtre d’émission. Ce signal est ensuite
9Figure 1.3 – Le produit de la surface de la source et son cône forme un invariant optique.[38]
capté par un détecteur matriciel comme une caméra CCD. Ainsi, tout le champ de vue est
acquis simultanément.
Malgré sa simplicité d’utilisation, la microscopie à champ large n’a pas de sectionnement
optique, ce qui signifie que la lumière provenant de tout l’échantillon est captée par le détec-
teur. La lecture devient alors difficile en raison du rapport signal sur bruit assez faible. C’est
pour cette raison que ce type de microscopie est plus adapté aux échantillons minces. Le sec-
tionnement optique provient du fait que le signal provient de quelques dizaines de micromètres
d’épaisseur de tissu.
1.7 Microscopie à balayage laser
La microscopie à balayage laser consiste à focaliser un faisceau laser en un point pour y
exciter des protéines fluorescentes. C’est en balayant ce point que l’on reconstruit une image
pixel par pixel. Ce principe est au coeur de la microscopie confocal qui est très répandue dans
les laboratoires de recherche. C’est un microscope à fluorescence qui utilise l’excitation mono-
photonique comme agent de contraste. La puissance de cette méthode provient de l’utilisation
d’un sténopé dans le parcours optique qui permet d’obtenir une résolution axiale de l’ordre
du micromètre. En effet, le sténopé va bloquer la lumière ne provenant pas du plan focal. En
utilisant des objectifs avec de grandes ouvertures numériques, il est donc possible d’obtenir
également une résolution latérale très fine, proche de la limite de Rayleigh.
Le même principe de balayage est utilisé dans la microscopie multiphoton. Cependant, les
sténopés ne sont plus nécessaires, puisque l’excitation multiphoton va se produire seulement
au point focal du faisceau laser, là où la probabilité que plusieurs photons soient absorbés en
même pour l’excitation est la plus élevée. C’est entre autres pour cette raison que les lasers
utilisés pour ce genre de microscopie utilisent des impulsions ultra-brèves à haute puissance
pour augmenter l’irradiance à l’illumination et ainsi les probabilités d’absorption.
10Vous pouvez aussi lire

















































