Matériel éducatif pour le médecin - AFMPS
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Les autorités de santé de l’Union Européenne ont assorti
la mise sur le marché du médicament Ruconest® de
certaines conditions. Le plan obligatoire de minimisation
des risques en Belgique et au Luxembourg, dont cette
information fait partie, est une mesure prise pour garantir
une utilisation sûre et efficace du médicament Ruconest®
(RMA version modifiée 11/2021).
RUCONEST® (conestat alfa)
2100 U, poudre pour solution injectable / poudre et solvant pour solution
injectable
Matériel éducatif pour le médecin
Il n’y a aucune expérience concernant l’utilisation de Ruconest chez les femmes
enceintes ou qui allaitent. Ruconest® n’est pas recommandé pendant la
grossesse ou l’allaitement sauf si le médecin traitant estime que les bénéfices
l’emportent sur les risques possibles.
Ce matériel ne contient pas toutes les informations. Pour une information complète, lisez
attentivement le RCP (en annexe) avant de prescrire et/ou d’utiliser et/ou de délivrer
Ruconest®. Le texte complet et actualisé de ce RCP est disponible sur le site www.afmps.be,
rubrique « NOTICE et RCP d’un médicament ».
V8.3 BE_fr Feb2022 1 de 12
RUCONEST® : ÉVALUATIONS IMMUNOLOGIQUES
GUIDE DESTINÉ AUX PRESCRIPTEURS
Résumé
Ruconest® (conestat alfa) est un analogue recombinant de l’inhibiteur de la C1 estérase
humain (rhC1-INH), produit à partir du lait de lapines transgéniques exprimant le gène
codant pour le C1-INH humain et est indiqué dans le traitement des crises aiguës
d’angioedème.
Comme pour tous les produits protéiques administrés par voie intraveineuse, des réactions
d’hypersensibilité ne peuvent pas être exclues. Aussi, certaines précautions doivent être
respectées avant d’instaurer un traitement par Ruconest® et lorsque des réactions
allergiques ou l’absence de réponse clinique sont observées à la suite d’un traitement par
Ruconest®.
Les précautions à prendre sont les suivantes :
i) Avant d’instaurer un traitement par Ruconest®, les antécédents médicaux de tous
les patients doivent faire l’objet d’une évaluation afin d’identifier si le patient a pu
développer des signes et symptômes suggérant une allergie possible aux lapins. Il
est possible que le patient ne ne souvienne pas d’une allergie découverte
antérieurement ou qu’il ne sache pas qu’il est allergique aux poils de lapin. Le
médecin doit poser les questions appropriées afin de déceler si le patient peut être
allergique aux poils de lapin. Les questions suivantes peuvent être posées :
a. Avez-vous été en contact avec des lapins dans le passé ?
b. Lors de contacts avec des lapins, avez-vous eu des démangeaisons, une
éruption cutanée, des éternuements, le nez qui gratte et qui coule, les yeux
rouges et qui piquent, de la toux, un essoufflement ou une respiration sifflante, ou
avez-vous ressenti une quelconque gêne ?
En cas de réponse affirmative à la deuxième question, des thérapies alternatives
sans produits issus de lapins doivent être envisagées.
ii) Il est important qu’une information détaillée sur les symptômes de l’hypersensibilité
et de l’allergie soit communiquée au patient par le médecin traitant, et de s’assurer
que le patient connaît l’importance d’alerter immédiatement un médecin en cas
d’apparition de ces symptômes.
iii) La consigne de signaler immédiatement toute réaction allergique aux lapins doit être
rappelée régulièrement.
iv) Lorsque Ruconest® est prescrit à un patient, vous devez lui remettre la carte patient
Ruconest®.
La décision quant à l’utilisation d’un traitement à domicile pour un patient particulier doit
être prise par le médecin traitant. On dispose de données limitées concernant l’utilisation
de ce médicament sous forme de traitement à domicile ou auto-administré.
Par ailleurs, nous vous rappelons l’existence d’un registre post-commercialisation dans
lequel les professionnels de santé sont encouragés à introduire les patients.
V8.3 BE_fr Feb2022 2 de 12Introduction
La substance active de Ruconest® (conestat alfa) est un analogue recombinant de l’inhibiteur
de la C1 estérase humain (rhC1-INH). La séquence d’amino-acides de la forme
recombinante est identique à celle du C1-INH endogène. Le conestat alfa est exprimé dans
les cellules des glandes mammaires des lapines et extrait du lait de lapines transgéniques
exprimant le gène codant pour C1-INH. Bien que le processus de purification du rhC1-INH
ait été conçu pour éliminer le maximum possible d’Impuretés Provenant de l’Hôte (IPH),
Ruconest® contient quand même des traces de protéines de lapin provenant du lait de
lapine.
Les protéines recombinantes telles que Ruconest® peuvent induire la formation d’anticorps
dirigés contre la protéine recombinante et son homologue endogène, ainsi que contre des
Impuretés Provenant de l’Hôte (IPH).
Comme pour toutes les protéines administrées par voie intraveineuse, des réactions
d’hypersensibilité ne peuvent pas être exclues. Avant l’instauration du traitement par
Ruconest®, les patients devront être interrogés pour rechercher une allergie connue ou
suspectée aux lapins : une exposition préalable aux lapins, signes et symptômes évoquant
une réaction allergique. Dans ce cas, des thérapies alternatives sans produits issus de lapins
doivent être envisagées. Les patients doivent être étroitement surveillés et
soigneusement examinés pour déceler tout signe et symptôme d’hypersensibilité
durant et immédiatement après les périodes d’administration. En cas de réactions
anaphylactiques ou de choc, un traitement médical d’urgence doit être administré.
Un traitement par Ruconest® doit être instauré sur les conseils et sous la surveillance
d’un médecin expérimenté dans le diagnostic et le traitement de l’angiœdème
héréditaire et il doit être administré par un professionnel de santé.
Les réactions immunologiques potentiellement associées à Ruconest® sont développées
dans trois chapitres :
1. Hypersensibilité de type I (réactions immédiate ou anaphylactique),
2. Anticorps neutralisants (à l’origine d’une efficacité réduite),
3. Hypersensibilité de type III (hypersensibilité liée aux complexes immuns).
Chacun de ces trois chapitres porte sur :
• Les mécanismes impliqués dans les réactions,
• Les faits et résultats étayant le risque,
• Les tests auxquels on peut recourir pour prévenir une telle réaction, et les tests
disponibles pour explorer les événements suspectés d’avoir une origine
immunologique,
• Le traitement.
V8.3 BE_fr Feb2022 3 de 121 HYPERSENSIBILITÉ DE TYPE I (réactions immédiates ou anaphylactiques)
Mécanisme : L’hypersensibilité de type I qui peut prendre la forme d’une urticaire, d’une
conjonctivite, d’une rhinite, d’une dyspnée et / ou d’un choc est médiée par des anticorps
IgE. Elle peut être difficile à distinguer d’une crise d’angiœdème héréditaire (AOH). Les
anticorps IgE sont induits par une exposition antérieure à un antigène identique ou similaire
à celui déclenchant la réaction.
Par exemple :
• Les patients allergiques au lapin peuvent avoir des anticorps IgE préexistants
susceptibles de réagir aux traces d’impuretés protéiques de lapin contenues dans
Ruconest®.
• Bien qu’une réactivité croisée entre le lait de vache et le lait de lapine soit considérée
comme peu probable, la possibilité d’une telle réactivité croisée chez un patient
présentant des signes d’allergie clinique au lait de vache ne peut être exclue et le
patient doit faire l’objet d’une surveillance pour déceler tout signe et symptôme
d’hypersensibilité suite à l’administration de Ruconest®.
Les faits : Dans le programme d’essais cliniques sur le Ruconest®, un volontaire sain a
présenté une réaction d’hypersensibilité de type I à la suite d’une première exposition au
Ruconest®. Le sujet avait une allergie préexistante au lapin, non révélée. Entre temps, plus
de 1 500 doses ont été administrées à 268 personnes dans le cadre d’essais cliniques sur le
Ruconest® sans qu’aucun autre cas d’hypersensibilité sévère/de réaction anaphylactique ne
soit signalé.
Antécédents médicaux : À cause du risque possible de réactions allergiques,
le Ruconest® est contre-indiqué chez les patients ayant une allergie au lapin
avérée ou suspectée.
Pour cette raison :
Avant d’instaurer un traitement par Ruconest®, les antécédents médicaux et
l’état de santé actuel de tous les patients doivent faire l’objet d’une évaluation
minutieuse afin de détecter toute allergie aux lapins connue ou suspectée.
Traitement : Les patients traités par Ruconest® doivent être surveillés pour
détecter d’éventuels signes cliniques et symptômes d’hypersensibilité durant et
juste après l’administration.
Un traitement médical d’urgence doit être directement disponible pour être administré
immédiatement en cas de réaction anaphylactique ou de choc.
Il est important d’informer les patients sur les signes précoces de réactions
d’hypersensibilité de type I, comprenant les éruptions urticariennes, l’urticaire généralisée,
l’oppression thoracique, la respiration sifflante, l’hypotension et l’anaphylaxie, et sur le fait
qu’ils doivent alerter immédiatement leur médecin en cas d’apparition de ces symptômes.
Les patients doivent également recevoir régulièrement la consigne de signaler toute réaction
allergique aux lapins (ex. annuellement).
Il est également important d’informer le patient sur les différences entre une crise aiguë
d’angiœdème héréditaire et une réaction d’hypersensibilité.
Si une hypersensibilité de type I au Ruconest® est suspectée ou si des symptômes d’allergie
se manifestent, le traitement par Ruconest® doit être arrêté. D’autres traitements sans
produits issus de lapins doivent être envisagés.
V8.3 BE_fr Feb2022 4 de 122 ANTICORPS NEUTRALISANTS (entrainant une diminution de l’efficacité)
Mécanisme : La synthèse d’anticorps neutralisants dirigés contre C1-INH peut réduire le
taux de C1-INH fonctionnel et avoir pour résultat une réponse clinique réduite (voir ci-
dessous les critères pouvant amener à recourir à des tests immunologiques). Si des
anticorps devaient se lier au C1-INH endogène, la situation clinique deviendrait comparable
à celle d’un angiœdème acquis (AOA) ; une éventualité rare, habituellement provoquée par
la présence d’anticorps neutralisants anti-C1-INH endogène produits par un lymphome ou
associés à une maladie auto-immune.
Les faits : Lors du programme d‘essais cliniques, des échantillons de plasma ont été
recueillis avant et après exposition au Ruconest®. La présence d‘anticorps anti-C1-INH dans
les échantillons plasmatiques, a été recherchée grâce à 6 dosages ELISA différents
(Enzyme Linked Immuno Sorbent Assays) qui détectaient les anticorps IgM, IgG et IgA
dirigés contre le pdC1-INH, inhibiteur de l’estérase C1 extrait du plasma (pdC1-INH pour
plasma-derived C1-INH) et dirigés également contre son recombinant rhC1-INH (les
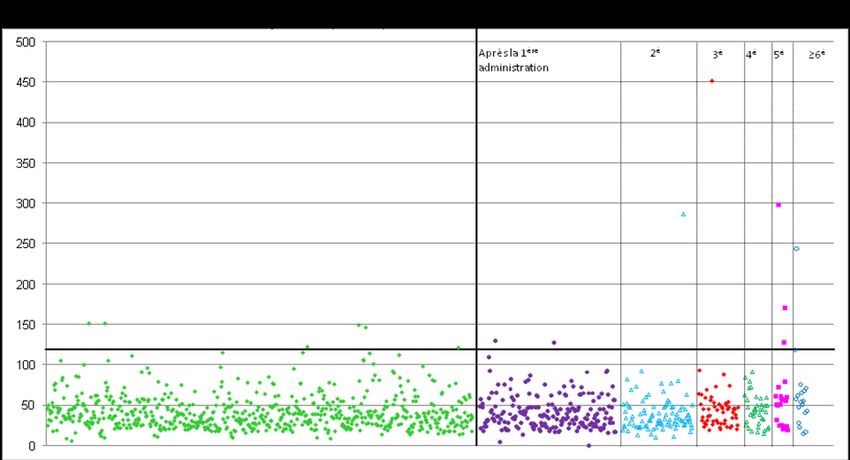
résultats des IgG sont montrés dans la figure ci-dessous).
Echantillons avant exposition (n=691) Echantillons après exposition (n=492)
ELISA résultat (AU)
La figure montre les mesures des anticorps IgG spécifiques anti-rhC1-INH chez des patients souffrant d’angiœdème héréditaire
avant le 1er traitement par rhC1-INH et après. La ligne rouge horizontale représente le seuil du dosage ELISA. La dernière
colonne est la compilation de tous les échantillons prélevés après exposition chez des sujets ayant reçu entre 6 à 26
administrations.
Les échantillons plasmatiques ayant des taux d’anticorps au-dessus du seuil ont été testés
pour les anticorps neutralisants anti-C1-INH. Les résultats se résument comme suit :
• Aucune réponse persistante pour les anticorps anti-pdC1-INH et anti-rhC1-INH n’a été
observée.
• Aucun anticorps neutralisant anti-C1-INH n’a été trouvé.
Tests : La présence d’anticorps neutralisants doit être suspectée devant une réponse
clinique réduite si :
(a) Lors de deux crises consécutives d’angiœdème chez des patients souffrant
d’angiœdème héréditaire qui répondaient précédemment à un traitement par
V8.3 BE_fr Feb2022 5 de 12Ruconest® à la dose de 50 U/kg, il s’est maintenant avéré nécessaire d’administrer
deux doses de Ruconest® pour traiter chacune des crises aiguës.
ET / OU
(b) Lors de deux crises consécutives d’angiœdème chez des patients souffrant
d’angiœdème héréditaire qui répondaient précédemment au traitement par Ruconest®
à la dose de 50 U/kg, il a été observé dans les 4 heures, un échec de la réponse au
traitement par Ruconest® à la dose standard de 50 U/kg.
Pour la première étape diagnostique, il est recommandé de mesurer l’activité fonctionnelle
du C1-INH 15 minutes après une perfusion de Ruconest® à la dose de 50 U/kg. Des
concentrations de C1-INH>0,7U/ml excluent la présence d’anticorps neutralisants,
cliniquement significatifs.
Pharming Technologies B.V. fournit un programme de tests immunologiques pour surveiller
l’émergence d’anticorps neutralisants que l’on suspecte. Le programme de tests doit être mis
en œuvre chez les patients souffrant d’un angiœdème héréditaire remplissant les critères (a)
ou (b) mentionnés ci-dessus, et qui n’atteignent pas un taux de C1-INH fonctionnel >0,7U/ml,
15 minutes après l’administration de Ruconest® à la dose de 50 U/kg (voir ci-dessus) :
Une trousse de tests peut être demandée par email à medinfo.be@pharming.com (pour la
Belgique) ou à medinfo.lu@pharming.com (pour le Luxembourg).
Le recueil des échantillons plasmatiques et les procédures d’expédition sont détaillés dans
l’ANNEXE A.
Les échantillons doivent être testés selon l’algorithme suivant :
• Des tests ELISA détectent la présence des anticorps IgG et IgM anti rhC1-INH. Ces
tests reposent sur la liaison de ces anticorps à un rhC1-INH immobilisé.
• Si les titres d’anticorps anti-rhC1-INH se situent au-dessus du seuil normal, on
confirme par un test de déplacement qui discrimine les réponses spécifiques et non
spécifiques.
• Si la présence d’anticorps spécifiques est confirmée, leur capacité à neutraliser un
pdC1-INH fonctionnel est testée par un dosage des anticorps neutralisants.
La stratégie complète du dosage des anticorps neutralisants suspectés est résumée dans le
protocole qui suit :
V8.3 BE_fr Feb2022 6 de 12Lors de deux poussées successives :
• Nécessite d’une dose >50 U/kg ET / OU
• Pas de réponse dans les 4h
C1-INH fonctionnel ≥ 0,7 U/ml Pas de suspicion d’anticorps
15 minutes après perfusion < 0,7 U/ml neutralisants
≤ seuil
lgG et IgM anti-rhC1-INH
> seuil
négatif
Test de déplacement
positif
Liaison, mais présence
d’anticorps
non-neutralisants*
Dosage des anticorps négatif
neutralisants positif
Anticorps neutralisants
confirmés
* Ces anticorps peuvent augmenter la clairance du rhC1-INH.
Traitement : Chez les patients ayant des anticorps neutralisants, le tableau clinique et les
options thérapeutiques sont les mêmes que chez les patients souffrant d’angiœdème acquis
(AOA). Une crise aiguë d’AOA nécessite généralement de plus fortes doses de C1-INH que
celles administrées dans la forme héréditaire. L’administration de C1-INH est le traitement de
choix lors des crises d’AOA menaçant le pronostic vital. Devant une telle situation, en plus
des gestes d’accompagnement tels que l’intubation en cas d’AOA menaçant le pronostic
vital, un traitement par icatibant peut être envisagé.
V8.3 BE_fr Feb2022 7 de 123 HYPERSENSIBILITÉ DE TYPE III (hypersensibilité aux complexes immuns)
Mécanisme : La synthèse d’anticorps anti-C1-INH ou anti-IPH (Impuretés Provenant de
l’Hôte) peut provoquer une hypersensibilité de type III. La réaction dont les intermédiaires
sont des complexes immuns peut se généraliser ou bien toucher un organe spécifique (en
témoignent les symptômes d’une « réaction transfusionnelle » ou d’une « maladie sérique »).
Les faits : Lors du programme d’essais cliniques, des échantillons de plasma prélevés avant
et après exposition, ont été recueillis. En plus du dépistage des anticorps anti-C1-INH décrits
précédemment, un test ELISA (Enzyme Linked Immuno Sorbent Assay) a été utilisé pour
détecter la présence d’anticorps IgM, IgG et IgA anti-impuretés provenant de l’hôte (les
résultats sont montrés sur la figure ci-dessous).
ELISA
La figure montre les estimations des anticorps anti-impuretés provenant de l’hôte, chez des patients souffrant d’un angiœdème
héréditaire symptomatique, après traitement répété par rhC1-INH. La ligne rouge représente le seuil du dosage ELISA. La
dernière colonne est une compilation d’échantillons après exposition provenant de sujets ayant reçu entre 6 à 26
administrations.
Les échantillons plasmatiques trouvés positifs lors du dépistage par tests ELISA, ont fait
l’objet d’un test de déplacement pour confirmation. Les résultats se résument comme suit :
• Aucune réponse persistante pour les anticorps anti-C1-INH n’a été observée.
• Des anticorps anti-impuretés provenant de l’hôte et dont les titres se situaient au-
dessus du seuil, ont été observés chez quelques patients ; mais ils n’étaient pas
associés à des symptômes cliniques de réaction immunologique.
Tests : Pharming Technologies B.V. fournit un programme de tests immunologiques pour
suivre les réactions d’hypersensibilité qui sont suspectées. Ce programme de tests doit être
envisagé chez les patients souffrant d’un angiœdème héréditaire (AOH) remplissant les
critères (c) et/ou (d) ci-dessous :
(c) Une réaction d’hypersensibilité de type III (avec des symptômes cutanés, articulaires
ou rénaux) qui survient dans les jours ou semaines suivant une administration de
V8.3 BE_fr Feb2022 8 de 12Ruconest®, et qui après investigation d’autres causes possibles, ne peut pas être
complètement expliquée par une exposition et une réaction aux autres antigènes que
ceux contenus dans Ruconest®.
(d) Des réactions d’hypersensibilité de type III observées à deux reprises consécutives,
dans les jours ou semaines suivant une administration de Ruconest®.
Une trousse de tests peut être demandée par email à medinfo.be@pharming.com (pour la
Belgique) ou à medinfo.lu@pharming.com (pour le Luxembourg).
Le recueil des échantillons plasmatiques et les procédures d’expédition sont détaillés dans
l’ANNEXE A.
V8.3 BE_fr Feb2022 9 de 12Les échantillons doivent être testés selon l’algorithme suivant :
• Des tests ELISA détectent les anticorps IgG et IgM anti-rhC1-INH. Ces tests reposent
sur la liaison de ces anticorps à un rhC1-INH immobilisé.
• Si les titres des anticorps anti-rhC1-INH se situent au-dessus du seuil, on confirme par
un test de déplacement qui discrimine les réponses spécifiques et non spécifiques.
• Un autre test ELISA détecte la présence des Ig totales dirigées contre les impuretés
provenant de l’hôte. Ce test ELISA dirigé contre les impuretés contenues dans un éluat
de phase solide (HRISP-eluate ELISA) mesure la liaison des anticorps aux antigènes
récupérés lors la première étape de purification du processus de fabrication du
Ruconest® (éluat de la phase solide). Ce test est par conséquent hautement spécifique
et sensible aux dites impuretés contenues dans le Ruconest®.
• Si des titres d’Ig totales situées au-dessus du seuil sont observés lors du test dirigé
contre les impuretés contenues dans l’éluat provenant de la phase solide, un test de
déplacement est effectué pour confirmation sur l’échantillon afin de faire la
discrimination entre les réponses spécifiques et non spécifiques.
Le protocole ci-dessous résume la stratégie complète du test de l’hypersensibilité de type III
au Ruconest®, qui est suspectée :
Réaction d´hypersensibilité de type III (peau, articulations ou reins) :
• événement unique non explique par d´autres antigènes
• et qui fait suite á deux administrations consécutives
Test des anticorps anti-
≤ seuil
impuretés contenus dans
Pas d´anticorps suspectés
l´éluat de la phase solide > seuil
négatif
Test de déplacement
positif
≤ cut-off
IgG et IgM anti-rhC1-INH
> cut-off
Anticorps confirmés
négatif
Test de déplacement
positif
Traitement : Les patients ayant des résultats positifs lors des recherches d’anticorps doivent
être exclus des traitements ultérieurs par Ruconest®. Le traitement symptomatique de courte
durée d’une hypersensibilité de type III comporte des agents anti-inflammatoires.
V8.3 BE_fr Feb2022 10 de 12Notification des effets indésirables Pour la Belgique : Les professionnels de la santé sont invités à notifier les effets indésirables ainsi que les éventuelles erreurs médicamenteuses liés à l’utilisation de Ruconest® à la division Vigilance de AFMPS. La notification peut se faire de préférence en ligne via www.notifieruneffetindeisrable.be, sinon à l’aide de la « fiche jaune papier » disponible sur demande à l’AFMPS ou imprimable à partir du site internet de l’AFMPS, www.afmps.be. La « fiche jaune papier » remplie peut être envoyée par la poste à l’adresse AFMPS – Division Vigilance – Avenue Galilée 5/03 – 1210 Bruxelles, par fax au numéro 02/528.40.01, ou encore par email à l’adresse adr@afmps.be. L’adresse de la boîte postale de l’AFMPS (Boîte postale 97, 1000 Bruxelles Madou) peut également être utilisée. Pour le Luxembourg : Les professionnels de la santé sont invités à notifier les effets indésirables liés à l’utilisation de Ruconest® à la Direction de la Santé - Division de la Pharmacie et des Médicaments, 20, rue de Bitbourg, L-1273 Luxembourg-Hamm , par e-mail à pharmacovigilance@ms.etat.lu ou par téléphone au numéro +352 247 85592 ou à Centre Régional de Pharmacovigilance de Nancy, Bâtiment de Biologie Moléculaire et de Biopathologie (BBB), CHRU de Nancy – Hôpitaux de Brabois, Rue du Morvan, 54 511 VANDOEUVRE LES NANCY CEDEX par e- mail : crpv@chru-nancy.fr ou par téléphone au +33 3.83.65.60.85 / 87. Lien pour le formulaire : https://guichet.public.lu/fr/entreprises/sectoriel/sante/medecins/notification-effets- indesirables-medicaments.html Les effets indésirables doivent être notifiés à Pharming Group N.V. par email à l’adresse safety.be@pharming.com (pour la Belgique) ou safety.lu@pharming.com (pour le Luxembourg). V8.3 BE_fr Feb2022 11 de 12
ANNEXE A
RECUEIL D’ÉCHANTILLONS DE PLASMA ET DES PROCÉDURES D’EXPÉDITION.
Sur demande, pour les patients susceptibles d’avoir développé des anticorps neutralisants
ou une hypersensibilité de type III au Ruconest®, et remplissant les critères spécifiés dans le
guide d’information, une trousse destinée aux tests d’évaluation immunologique du
Ruconest® sera envoyée avec le contenu suivant :
• 8 étiquettes
• 2 tubes citratés
• 4 cryo-flacons
• Instructions pour le recueil des échantillons de plasma (voir ci-dessous)
• Fax de notification du recueil des échantillons
INSTRUCTIONS POUR LE RECUEIL DES ÉCHANTILLONS DE PLASMA :
Avant de prélever un échantillon de sang, indiquer sur l’étiquette les renseignements
concernant le patient et la date.
• Prélever 4,5 ml de sang sur un tube citraté par une technique de ponction veineuse.
• Etiqueter le tube citraté avec l’étiquette d’identification du patient posée
horizontalement, et non sur la longueur du tube.
• Centrifuger l’échantillon pour séparer le plasma des hématies à 1000 x g (soit
environ 2000 tpm) pendant 10 à 15 minutes, à la température de la pièce.
• Etiqueter les cryo-flacons avec l’étiquette d’identification du patient posée
horizontalement.
• Retirer le plasma (ici le surnageant) et transférer 2 x 0,75 ml de plasma dans des
cryo-flacons.
• Congeler et stocker les échantillons de plasma verticalement (2 cryo-flacons x 0,75
ml) à ≤−70 °C dès que possible.
• Jeter le plasma restant conformément à la règlementation en vigueur.
EXPÉDITION DES ÉCHANTILLONS DE PLASMA :
Envoyer un fax de notification du recueil des échantillons ; le coursier vous contactera alors
pour organiser la récupération des échantillons.
V8.3 BE_fr Feb2022 12 de 12Vous pouvez aussi lire

















































