Chorée - diagnos/c et traitement - Anna HEINZMANN, MD Département de Géné5que & ICM, Hôpital de la Pi5é-Salpêtrière, Paris - Movement Disorder Society
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous

Chorée - diagnos/c et traitement
Anna HEINZMANN, MD
Département de Géné5que & ICM,
Hôpital de la Pi5é-Salpêtrière, Paris
Chorée - diagnos/c et traitement
Diagnos/c différen/el de la chorée
• Causes acquises
• Causes géné5ques
• Maladie de Hun5ngton
• Autres causes géné5ques
Prise en charge (modèle de la maladie de Hun5ngton)
• Traitement
• Pistes thérapeu5ques
• Test présymptoma5que
68 year old male, history of lung 20 year old women, no medical history cancer 10 years ago, in remission, subacute chorea Chorée paranéoplasique (an/-CV2) Chorée avec an/corps an/phospholipides Remerciements J. Honnorat et E.Roze
Aquired choreas
Autoimmune disorders Metabolic
Sydenham: TOC, hyperac5vité, aXeinte cardiaque, arthrite Hyperglycemia, hyper/hyponatremia
Lupus/an5phospholipide aXeinte systémique? nutri5onal (B12)…
Paraneoplas5c cancer?
hormonal Drugs/toxins
Contracep5on, pregnancy
Hormone replacement therapy, Levodopa, agonistes de la dopamine,
thyroid dysfunc5on An5epilep5cs, cocaine, mercure, manganèse…. Zheng, fronBers neurol 2020
hematological Structural basal ganglia lésions
polyglobulia Mass lesions, vascular lesions… unilateral
infec5ous
HIV…
Cardoso, lancet 2006 Penday, J Clinic Neuroscience, 2013
Basic workup
Blood work up
NFS, ionogramme, VS, CPK, FAN, an5phospholipides, an5-ADN, an5corps
an5neuronaux, T4-TSH, B12, B1, an5corps an5thyroidiens, glycémie,
acanthocytes, VIH, TPHA VDRL, bilan cuprique, bilan mar5al, phénotypage et
5tre an5gène Kell, AFP….
Brain imaging: look for
• lesions
• Atrophy paXern
• Caudate atrophy?
• Cerebellar atrophy?
• Brain stem atrophy?
• White maXer changes
• Iron accumula5on
2
1
3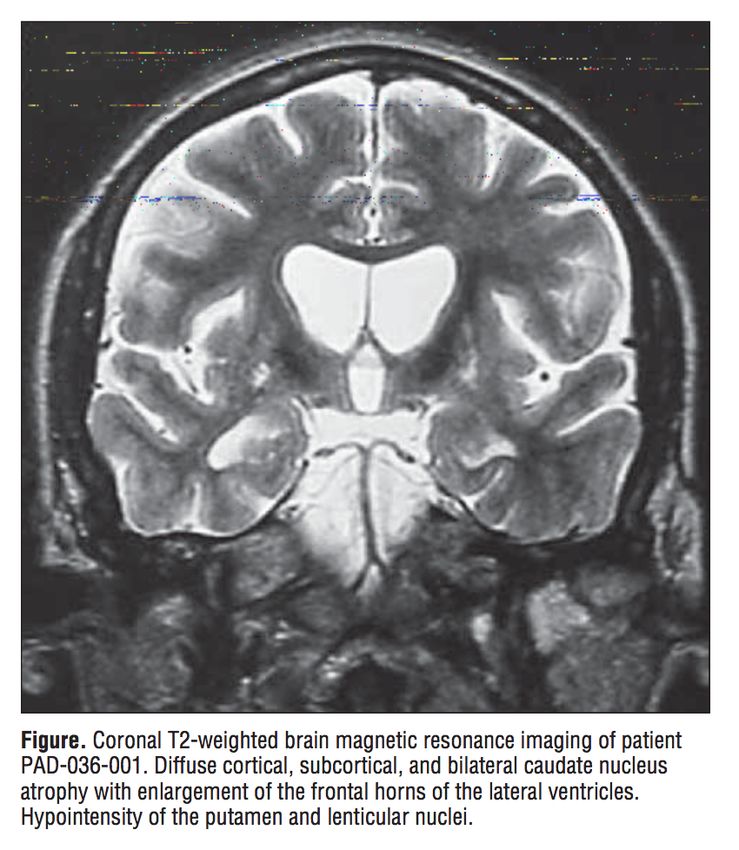
AXeinte motrice variable et trouble de la marche polymorphe: chorée + dystonie, extrapyramidal, stabilité posturale, éléments cérébelleux, dysarthrie, troubles oculomoteurs, aXeinte pyramidale…

Hun/ngton’s disease (HD)
• Autosomal dominant, neurodegenera/ve, leads to death in 15-20
years
• CAG expansion in the HTT gene (IT15) coding for Hun5ng5n,
localised on the short arm of chromosome 4 (4p16.3), iden5fied in
1993
36-250
6-35 4p16.3
exon1
• Hun5ng5n expression ubiquitous, mul5ple func5ons
• HTT knock out in adults is followed by neurodegenera5on
• HTT knock out in the embryo is lethal
HD – a neurodevelopmental disease?
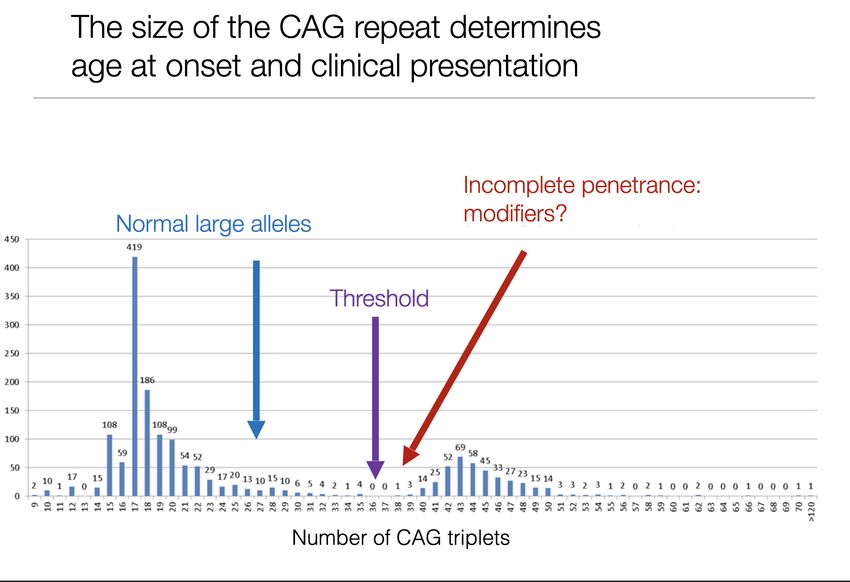
HD – prevalence,penetrance • Prevalence : 4-10/ 100 000 (Occident) • Onset between 35-50, 5-10% before 20, 30% ajer 50 • age dependent penetrance,complete > 39,can be incomplete for 36-39 CAG
age at onset depends on the size of the CAG expansion
Lee et al 2012
• CAG size determines 50-70 % of variability of age at onset
• Age at onset more variable for small expansionsGene/c modifiers of age at onset – not (yet?) used in clinical rou/ne
GeM-HD consortium, Cell 2015
• CAG repeat and not polyglutmine lenght determines age of onset
• Important influence in the reduced penetrance range (small expansions)
• Age of onset almost 10 years earlier than es/mated in variant carriers
Wright et al 2019, Hayden et al 2020, Black et al 2020Symptoms (motor)
Classique - triade
• ATeinte motrice
• AXeinte cogni5ve
• AXeinte psychiatrique/troubles du
comportement
• + perte de poids
• Au début pas de mouvements choréiques
• Mais troubles du comportement/cogni5fs
discrets aspécfiques (irritabilité, lenteur,
difficultés de concentra5on)
• Troubles psychiatriques (anxiété, dépression)
UHDRS: score
moteur: X/124
Meilleur score = 0
Maximal pire = 124Pre- Pre-
Symptomatic
lesion manifest
UHDRS: score moteur: X/124
Meilleur score = 0, valeur max pire: 124
Score de confidence (DCL):
DCL 0 = normal
1=99% confidence
Manifest HD
Clinical signs
Prodromal phase
Presymptomatic phase
Disease
Pathological onset
Genetic predisposition
TimeSymptoms (cogni/on)
Classique - triade
• Absence de défini/on formelle de la démence/
• AXeinte motrice
MCI dans la MH
• ATeinte cogni/ve
• AXeinte psychiatrique/troubles du • Anosognosie
comportement • bradyphrénie+++
• + perte de poids • Apathie
• AXeinte dysexécu5ve
• AXeinte de la cogni5on sociale/ théorie de
l'esprit
• Troubles aXen5onnelsSymptoms (Psychiatric and behavioural)
• Behavioural disorders:
Classique - triade persevera5ons, behavioural
• AXeinte motrice deshinibi5on, irritability
• AXeinte cogni5ve • apathy
• ATeinte psychiatrique/troubles du • Psychiatric disorders with very
comportement frequent depression, addic5ons
• + perte de poids • anxiety
• Suicide rate higher than the general
popula5on???
Hyposignal putaminal T2
Atrophie du noyaux caudé
Remerciements Perrine CharlesJuvenile Hun/ngtons Disease
• Defini5on: onset before the age of 20
• CAG approx. 65, an5cipa5on++ (paternal)
• 5% of HD pa5ents
• bradykinesia/rigidity/dystonia (chorea)
• developmental delay or cogni5ve impairment
• Epilepsy
• Behavioural disorders
• Faster progression, earlier death
Fussili et al 2018Therapy –motor symptoms
Aucun traitement cura5f/neuroprotecteur efficace
Chorea:
• Tetrabenazine
• Neurolep5cs
• atypical: Olanzapine/Zyprexa,
Apipiprazol/Abilify, Risperidone/
Risperdal
• typical: Tiapride/Tiapridal)
• anxioly5c, effec5ve on behavioural
symptoms
• Physiotherapy+++
With neurolep/cs you can help HD pa/ents twice:
« Once when you start them, again when you stop them »
(Shoulson, Neuropharmacol, 1986)
ATen/on pas de traitement « cosmé/que »Therapy –non motor symptoms
• Psychiatric symptomes:
• An5depressants
• anxiolyiques
• Neurolep5cs
• Psychiatrist/psychologist
• Cogni/ve symptoms:
• Speech and language therapy
• Weight loss:
• Dietary supplements
• In general:
• Social supporttreatment perspec/ves in 2021
Bates et al. 2015 Nat Rev.treatment ? an/sense oligonucleo/des (ASO)
Principe des ASO dans la MH
gain of (toxic) func5on -> gene silencingtreatment ? an/sense oligonucleo/des (ASO)
Durr. Medecine Science 2019ASO: allèle spécifique
Wave life science, PRECISION HD trialTest présymptoma/que: forme contemporaine d’un oracle?
Tests présymptoma5ques des maladies neurogéné5ques La Salpêtrière 1992-2017
Autres,
CMT
ALZ
CJD 1% Huntin
SPG
gton
SCA
Disease
, 1 967Test présymptoma/que « c’est arrivé demain »
Test présymptoma/que-les enjeux
• Gravité de l’affec5on
• Possibilité de traitement cura5f ou de préven5on – non pour la MH
• Résultat binaire
• porteur vs non porteur
• mais complexe (persistance de mul5ples incer5tudes: pénétrance complète/
incomplète/âge dépendante, expressivité variable
• Condamna5on vs libéra5onTest présymptoma/que: un disposi/f mul/disciplinaire
Demande de la personne à risque
♦ Phase d’information (généticien)
Le temps pour réfléchir et décider
♦ Temps de réflexion
Abandon
(psychologue, assistante sociale,
temporaire
généticien, psychiatre) ou définitif possible
Décision,
♦
à tout moment
prélèvement sanguin et analyse
♦ Rendu du résultat
♦ Phase de suivi pluridisciplinaireLes faits marquants du diagnos/c présymptoma/que des maladies
neurologiques à révéla/on tardive
• Les demandes sont rares et ne progressent pas (es5ma5on: 5 to 25% des personnes à
risque)
Hayden Lancet 2000, Goizet et al Neurology 2002, Tassicker et al Eur J Hum Genet 2008, Morrison et al Clinical GeneBcs 2011
• 20 % des personnes à risque formulent une demande de test, 50% abandonnent après le
premier entre5en, 10% abandonnent après Goizet et al Neurology 2002
• Consulta5on pour test présymptoma5que à 35 ans environ (les enfants sont déjà là!)
• Mo/va/on variable selon la pathologie (HD: savoir, informer les enfants, SCA: crainte
d’être aXeinte)
• Diagnos/c prénatal est peu fréquent après un résultat défavorable (18% pour la porteurs
MH)
Gaudebout et al ESHG 2008
• effets indésirables graves limités:
• dépression fréquente chez le porteurs (58%) de la muta5on mais aussi chez les non porteurs
(24%)
• Facteur prédic5f pour la dépression après le test: la dépression avant le test Gargiulo et al EJMG 2003
• Non porteurs/résultat favorable:
• guérir du fait d’être à risque (le statut « d’être à risque » fait par5e de son iden5té)
• culpabilité du survivant par rapport à d’autres membres de la familleHun/ngton phénocopies - géné/ques
1-10% 3%
90-99%Hun/ngton phénocopies – géné/ques 1-10%
Schneider et al 2016???
Remerciements Louise Laure
Mariani et Perrine CharlesJPH3 (HDL 2)
• Repeat expansion disease (CAG/CTG)
• Autosomal dominant
• African origin+++
• nega5ve correla5on between age of onset and repeat length as in HD
• An5cipa5on
• Tableau clinique et paraclinique (paXern d’atrophie à l’IRM) comme la maladie de
Hun5ngton
+/- AcanthocytosisC9orf 72 – DLFT and ALS…HDL?
SLA Sclérose
latérale
amyotrophique
Hensman Moss et al 2014:
• Cohort of 514 HD phenocopy pa5ents
• 10/514 (1.95%), 3 with chorea
Motor neuron signs?
DFT Démence
frontotemporaleAtaxia genes
SCA 17 AOA
….
DRPLA
AT
CACNA1A
….
récessif
STUB1
dominantTBP/ SCA 17 (HDL 4)
• CAG/CAA expansions in the TBP gene encoding TATA-box-binding protein, a
transcrip5onal regulator
• AD transmission
• Ataxia + cogni/ve decline + psychiatric symptoms + chorea (20%)/dystonia +
epilepsy + pyramidal
Ataxia
epilepsy
cerebellar atrophy on MRI imaging
Rim enhancement of the putamen
Wild et al 2007ATN1/ DRPLA
• CAG expansion in ATN1
• AD transmission
• Japan+++ , rare in caucasians
• Ataxia + myoclonic epilepsy + chorea + cogni5ve decline+ psychiatric symptoms,
dystonie, sd parkinsonien
Ataxia
Myoclonic epilepsy (juvenile presenta5on+)
Cerebellar and brainstem atrophy on MRI imaging
hyperintense cerebral white maXer T2 lesions???
• 67 year old women
• Mild chorea (UHDRS 25/124)
• Mild ataxia (SARA 6/40)
• Cogni5ve complaint (execu5ve dysfunc5on, aXen5on deficit)
• Censured family history
• No progression
• No expansion in HTT (HD), JPH3 (HLD2), ATN1 (DRPLA), TBP (SCA17)
Absence of caudate atrophy, important cerebellar atrophyCACNA1A-related disorders
3 different well known allelic disorders
• familial hemiplegic migraine type 1 (FHM1)
• episodic ataxia type 2 (EA2) + Cogni5ve deficits and psychiatric
• spinocerebellar ataxia type 6 (SCA 6) - expansion Overlap++ symptoms increasingly recognized
But also
• Mental retarda5on/au5sm spectrum disorder
• Epilep5c encephalopathy
28 phenocopies (198 HTT expansions)
10 pa5ents: Inden5fica5on of the causa5ve gene (35%)
2 CACNA1A???
• 54 years old women
• Onset at 37 years: progressive cerebellar syndrome, chorea, cogni/ve
decline
• AD transmission (mother)
• SCA 1,2,3,6,7, TBP (SCA 17), ATN1(DRPLA), HTT, JPH3 (HLD2), C9orf72, PRNP
eliminated
Cerebellar atrophySTUB 1
STUB1 varia/ons (spinocerebellar ataxia type 16 (SCAR16)
➩ screening of 441 SCA families
➩ 7% (29/441) !
21/40 (53%) presented with cogni5ve impairment
HD-like (+ataxia)
FTLD-like (+ataxia)Remerciements M.Vidhailet
Acanthocytosis syndromes
Chorea acanthocystosis (VPS13A) Mc Leod Syndrome (XK)
• Autosomal recessive • X linked inheritance
• Extrapyramidal features,
cogni5ve decline, psychiatric
symptoms
• Progressive caudate atrophy
• Dystonia with prominent orofacial • Senorimotor neuropathy with
involvement areflexia
• Myopathy • Cardiac manifesta5ons
• Neuropathy • Epilepsy
• epilepsy
• Elevated CK • Nega5ve tes5ng for Kell an5gens
• acanthocytosisNeurodegenera/on with brain iron accumuma/on
Neuroferrinopathy (FLC gene) NBIA 1 (PKAN)
• Autosomal dominant Autosomal recessive
ac5on-specific facial dystonia (tongue pigmentary re5nal degenera5on
protrusion) Acanthocytosis
Low serum ferri5n eye of the 5ger sign
iron accumula5on in the basal ganglia
Wild et al 2007Conclusions
• First eliminate acquired chorea
• Most frequent gene5c causes: Hun5ngton Disease and HDL 2 (JPH3) in Africa
• Gene5c choreas: allelic and non allelic heterogeneity
• 1-10% of pa5ents with a HD-like phenotype with no CAG repeat expansions in
the HTT gene
Adult HD
Allelic heterogeneity (phenotype) Juvenile HD
Formes frustres
HD (HTT)
Ataxia genes
acanthocytosis
Non allelic heterogeneity
NBIA
HDL1
….Conclusions
Clinical signs
• Signes extraneurologiques?
• Signes neurologiques associés:
Clinical
characteris/cs
Bucco-
Re5nal Motor Ataxia
Epilepsy Neuropathy orolingual
degenera5on neuron
distribu5on
neurocanthocytosis
Neuroferrinopathy
orthers, DRPLA,
acanthocytosis
Acanthocytosis
SCA 17 and
(CACNA1A,
AT, AOA,
SCA17
AOA, AT
C9orf72
DRPLA
PKAN
STUB)
PKAN
Brain imaging
• Atrophy paXern
• Brain stem? Blood work up
• Caudate?
• Cerebellum?
• White maXer lesions
• Iron accumula5on in basal ganglia
• ………SUBJECT 503404 CIC, ICM
Merci pour votre
aTen/on
THE DAY WHEN MY HTT
WENT DOWNVous pouvez aussi lire

















































