ET PRISE EN CHARGE DES EI/EIG - RÉGLEMENTATION - Recherche Clinique Paris Descartes Necker | Cochin
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous

RÉGLEMENTATION
ET PRISE EN CHARGE DES EI/EIG
VIGILANCE DES ESSAIS CLINIQUES
DIU Formation des infirmières/techniciens
et chefs de projet
en recherche clinique
Sessions du 22 février 2019
et du 8 Mars 2019
Mylène TISSEYRE
Centre Régional de Pharmacovigilance
Service de Pharmacologie, Hôpital Cochin, AP-HP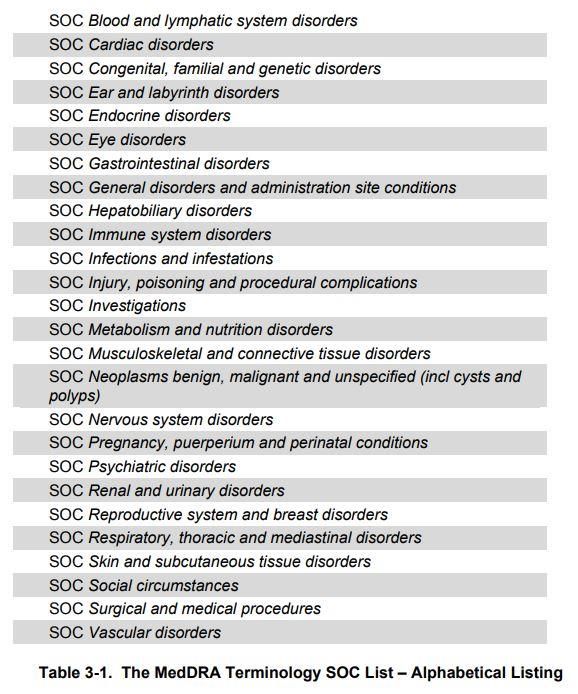
Plan
• Introduction : le système de pharmacovigilance en quelques
mots
• Vigilance des essais cliniques
– Cadre réglementaire
– Définitions
– Responsabilités de l’investigateur
– Responsabilités du promoteur
• En bref…
2
Pourquoi la pharmacovigilance?
Quelques dates…
• 1848 : Chloroforme et décès fortuit
• 1906 : US Federal Food and Drugs Act
• 1937 : Diéthylène glycol et sulfanilamide
• 1961 : Thalidomide et phocomélie (1962: amendements US
Federal Food and Drugs Act)
• 1964 : Yellow Card (UK)
• 1967 : Système de détection d’EI par l’OMS
• 1973 : Système de détection d’EI en France
• 1974 : practolol et syndrome oculo-muco-cutané
Routledge 1998 The Lancet 3
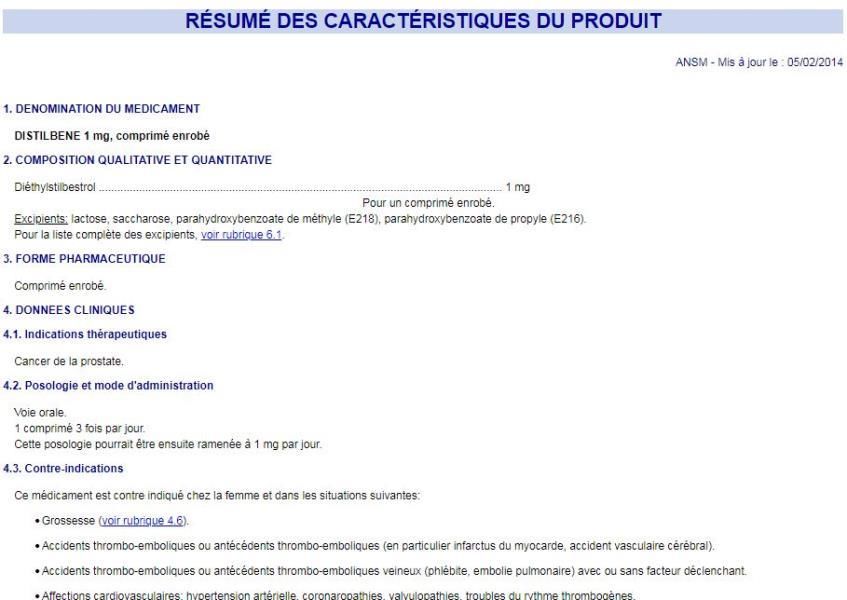
Pourquoi la pharmacovigilance? Cas du Distilbène® (diéthylstilbestrol (DES)) • 1948/1977 : en prévention des avortements spontanés, hémorragies gravidiques et diabète gestationnel • 1971 : 1ers cas de cancers du vagin chez des jeunes patientes exposées in utero au DES • 1977 : ajout de la contre-indication d’utilisation chez la femme enceinte • Aujourd’hui : AMM du Distilbène® cancer de la prostate Pour plus d’informations : https://www.ansm.sante.fr/var/ansm_site/storage/original/application/02c045c68 4fd4c49464654ff1349248d.pdf (consulté le 14/02/2019) 4
Pourquoi la pharmacovigilance?
Encore quelques dates :
• 1982 : benoxaprofène et photosensibilité/hépatotoxicité
• 2004 : Vioxx® (rofécoxib) et EI cardiovasculaires
• 2009 : Mediator® (benfluorex) et valvulopathies
Pour plus d’informations :
https://ansm.sante.fr/S-informer/Points-d-information-Points-d-information/Benfluorex-
Mediator-bilan-du-suivi-de-pharmacovigilance-Point-d-Information (consulté le
14/02/2019)
• 2015 : Dépakine® et effets tératogènes
• …
5
Du côté des essais cliniques
Essai TeGenero : TGN1412 – Londres, mars 2006
« Le premier essai humain d’un futur médicament tourne mal à Londres »
6
« First in Man » Source : http://www.hugoperen.org/reponses-des-questions-frequentes/ consulté le 23/03/2016 7

Du côté des essais cliniques
Essai TeGenero : TGN1412 – Londres, mars 2006
• 1er essai sur des humains anticorps monoclonal CD28-Super MAB
• Avril 2005 : feu vert de l’EMA ; juillet 2005 : feu vert du MHRA pour le
premier essai clinique dit de phase I
• Mais « cette autorisation se fondait sur les résultats des études menées
sur l'animal, laissant penser que ce produit était de nature à pouvoir
être utilisé contre des processus pathologiques de nature infectieuse,
immunitaire ou cancéreuse. Mais cette agence soulignait aussi que les
propriétés alors alléguées par le fabriquant allemand ne reposaient pas
sur des bases très claires. »
• Résultat : sur 8 volontaires sains, 6 sont hospitalisés en service de
réanimation ; les 2 volontaires indemnes sont ceux qui ont reçu le
placebo.
• Conséquences : changements réglementaires.
http://www.lemonde.fr/planete/article/2006/03/17/le-premier-essai-humain-d-un-futur-medicament-tourne-mal-a-
londres_751759_3244.html consulté le 23/03/16
8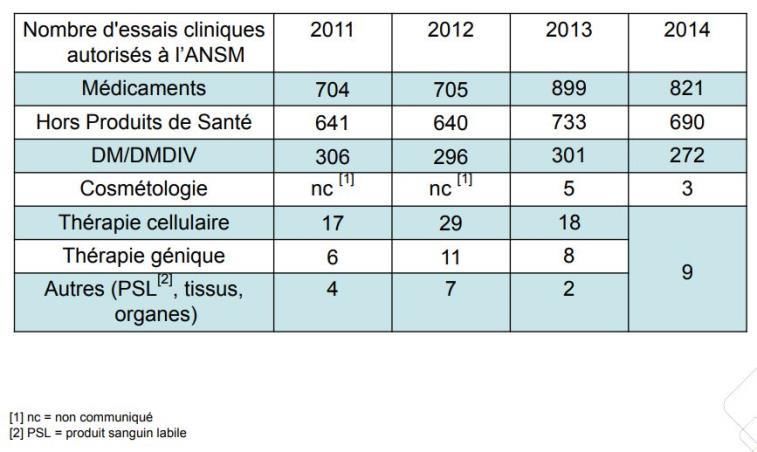
A quoi sert la pharmacovigilance?
Ensemble de techniques pour :
• Identifier / recueillir les EI des médicaments une fois sur le
marché
• Evaluer ces EI et les risques liés (notamment rapport
bénéfice/risque)
• Prévenir / informer / communiquer sur les risques potentiels
ou avérés
9
La pharmacovigilance…et les autres vigilances
8 vigilances (Code Santé Publique) - ANSM
• Pharmacovigilance : médicaments à usage humain et matières
premières à usage pharmaceutique
• Pharmacodépendance ou addictovigilance : substances psychoactives
dont stupéfiants et psychotropes
• Hémovigilance : toute la chaîne transfusionnelle (donneur suivi
receveur post-transfusion)
• Matériovigilance : dispositifs médicaux
• Réactovigilance : dispositifs de diagnostic in vitro
• Biovigilance : toute la chaîne de greffe (donneur suivi post-greffe
receveur) d’organes, tissus, cellules d’origine humaine (sauf sang et
gamètes)
• Cosmétovigilance : produits cosmétiques
• Vigilance des produits de tatouage
10La pharmacovigilance…et les autres vigilances Vigilances également au niveau de l’ANSES* • Nutrivigilance: compléments alimentaires et certains produits alimentaires • Phytopharmacovigilance : produits phytopharmaceutiques • Pharmacovigilance vétérinaire : médicaments vétérinaires • Toxicovigilance : effets toxiques (produit, substance ou pollution) *Agence Nationale de Sécurité Sanitaire de l’alimentation, de l’environnement et du travail https://www.anses.fr/fr/content/la-veille-et-la-vigilance consulté le 14/02/2019 11
Systèmes de vigilances
• Dates de mise en place différentes selon les vigilances
• Concourent toutes au même objectifs :
– exercer une surveillance de la sécurité d’emploi, du bon usage
– diminuer et prévenir les risques liés à leur utilisation par la mise
en place d’actions correctives ou préventives
assurer la sécurité du produit pour renforcer la sécurité des
personnes (patient, donneur ou utilisateur)
https://www.ansm.sante.fr/Declarer-un-effet-indesirable/Systemes-de-vigilances-de-l-Agence/Systemes-
de-vigilances-de-l-Agence/(offset)/0 consulté le 14/02/2019
12Comment fonctionne le système national de
pharmacovigilance?
Organisation en réseau
avec une coordination centrale
EMA
Système centralisé (activité de décision)
ANSM
CRPV Système décentralisé (activité de terrain)
CRPV = Centre Régional de Pharmacovigilance ; n =31 13Les essais cliniques en France - Quelques chiffres https://www.ansm.sante.fr/var/ansm_site/storage/original/application/d02d0b5b63ada81c5b1181c24226 d642.pdf consulté le 14/02/2019 14
Définitions
(article R1123-46 du CSP)
• Evènement indésirable : toute manifestation nocive survenant chez une
personne qui se prête à une recherche impliquant la personne humaine
que cette manifestation soit liée ou non à la recherche ou au produit sur
lequel porte cette recherche.
• Effet indésirable : évènement indésirable survenant chez une personne qui
se prête à une recherche impliquant la personne humaine, lorsque cet
évènement est lié à la recherche ou au produit sur lequel porte cette
recherche.
Définitions valables pour un médicament expérimental, un produit de
santé ou recherche ne portant pas sur un produit de santé
15Définitions
(article R1123-46 du CSP)
Critères de gravité d’une évènement ou d’effet indésirable EIG :
• Décès
• Mise en danger de la vie du patient
• Hospitalisation ou prolongation de l’hospitalisation
• Incapacité/handicap important ou durable
• Anomalie/malformation congénitale
16Définitions
Critères de gravité d’une évènement ou d’effet indésirable EIG :
• Tout évènement jugé comme médicalement important (IME)
• 6ème critère de gravité défini par l’ICH (International Conference on
Harmonisation)
• IME list = liste de référence
– Accès libre sur internet (site de l’EMA)
– Contient un peu plus de 8000 termes (dernière version) dont
infections/inflammations sévères, tumeurs, plupart des anomalies
congénitales/génétiques
17Définitions
(article R1123-46 du CSP et décret d’application loi Jardé du 16 novembre 2016)
Effet indésirable inattendu (SUSAR en anglais (Suspected Unexpected
Serious Adverse Reaction)) :
= tout effet indésirable dont la nature, la sévérité, la fréquence ou
l’évolution ne concorde pas avec les informations relatives aux produits,
actes pratiqués et méthodes utilisées au cours de la recherche.
Résumé des Caractéristiques du Produit (RCP)
Essai clinique Notice d’instruction/utilisation du DM/DMDIV
Brochure Investigateur (BI)
Agences réglementaires
Industrie pharmaceutique
(ANSM/EMA)
18Exemple de RCP
19Exemple de RCP – section 4.8
20Définitions
(article R1123-46 du CSP)
• Fait nouveau de sécurité (FNS) : toute nouvelle donnée pouvant conduire
à une réévaluation du rapport des bénéfices et des risques de la recherche
ou du produit objet de la recherche, à des modifications dans l’utilisation
de ce produit, dans la conduite de la recherche , ou des documents relatifs
à la recherche, ou à suspendre ou interrompre ou modifier le protocole de
la recherche ou des recherches similaires
NB: Pour les essais portant sur la première administration ou utilisation
d'un produit de santé chez des personnes qui ne présentent aucune
affection = tout effet indésirable grave. (loi Jardé)
21Définitions
• Promoteur
– Personne physique ou morale
– Prend l’initiative du projet
– En assure la gestion et/ou le financement
– Peut être institutionnel ou industriel
• Investigateur
– Personne physique
– Dirige et surveille la réalisation de l’essai sur un site
– Médecin
22Comment débute un essai clinique?
Avis
CPP favorable
Promoteur Début de
N° unique l’essai
dossier clinique
ANSM Autorisation
Base de données
européenne
(EVCTM* ; XEVMPD**)
* EudraVigilance Clinical Trial Module
** eXtended EudraVigilance Medicinal Product Dictionary 23Cadre réglementaire
Recherche portant sur les médicaments
• Directive 2001/20/EC du 4 avril 2001 relative à « l’application des
bonnes pratiques cliniques dans la conduite d’essais cliniques de
médicaments à usage humain »
• Directive 2005/28/EC du 8 avril 2005 relative à « l’application de
bonnes pratiques cliniques en ce qui concerne les médicaments
expérimentaux à usage humain »
• Loi n°2004-806 du 9 août 2004 relative à la politique de santé
publique et son décret d’application n°2006-477 du 26 avril 2006
(recherche biomédicale)
• Loi n°2012-300 du 5 mars 2012 (loi Jardé) relative aux recherches
impliquant la personne humaine et son décret d’application n°2016-
1537 du 16 novembre 2016
24Cadre réglementaire
Recherche portant sur les médicaments
• Au sujet de la Directive 2001/20/EC :
• Regulation (EU) n° 536/2014 du 16 avril 2014 relatif aux essais
cliniques de médicaments à usage humain
• Abroge la Directive 2001/20/CE
• Approche basée sur le risque
• Phase pilote en cours
• Mise en application attendue courant 2019
25Recommandations/guides
Recherche portant sur les médicaments
• CT-3 (June 2011) : detailed guidance on the collection, verification
and presentation of adverse event/reaction reports arising from
clinical trials on medicinal products for human use
• ICH guideline E6 (July 2002) : guidance on Good Clinical Practices ;
currently E6 (R2) (December 2016)
• ICH guideline E2F (September 2010) : guidance on Development
Safety Update Reports (DSUR)
• ICH guideline E2B (2003) : implementation guide on clinical safety
data management: data elements for transmission of Individual Case
Safety Reports (ICSRs) ; currently : E2B (R3) (juillet 2013)
• Avis aux promoteurs : mise en place et conduite en France d’essais
cliniques portant sur des médicaments à usage humain
26Recommandations/guides
Recherche portant sur les dispositifs médicaux
• Directive 93/42/CEE du 14 juin • Regulation
1993 relative aux dispositifs
(EU) n°
médicaux (DM)
2017/745 du
abroge 5 avril 2017
• Directive 90/385/CEE du 20 juin
1990 relative aux dispositifs relatif aux
médicaux implantables actifs DM
(DMIA)
• Directive 98/79/CE du 27 octobre • Regulation (EU) n°
abroge
1998 relative aux dispositifs 2017/746 du 5
médicaux de diagnostic intro vitro avril 2017 relatif
(DM-DIV)
aux DM-DIV
• Guidelines on a Medical Devices
Vigilance System MEDDEV 2.7/1
Rev 4 (juin 2016) & MEDDEV 2.7/3
Rev 3 (may 2015)
27En pratique que change la loi Jardé?
Loi de santé publique Loi Jardé
Recherche Recherche
biomédicale interventionnelle
Recherche
Recherche en
interventionnelle à
soins courants
risques minimes*
Recherche non Recherche non
interventionnelle interventionnelle
28Qu’est-ce qu’un médicament expérimental?
• Traitement testé
• Traitement comparateur
• Placebo
Directive 2001/20/EC
Un médicament expérimental n’est pas :
• Un médicament de secours
• Un médicament pour créer un état physiologique
• Un médicament utilisé pour un critère de jugement
• Un traitement concomitant
Directive 2001/83/EC et Regulation n°726/2004
29Médicament expérimental
(rôle du promoteur)
• Elaboration du dossier du médicament expérimental (ME)
• Veille à la fabrication, l’importation, l’approvisionnement, la
distribution, la gestion, la détention et la dispensation des ME
• Mise en place d’un système de rappel des ME
• Elaboration d’une procédure d’urgence de levée d’insu (selon le
protocole)
• Mise à disposition gratuite du ME
30Responsabilités de l’investigateur
31Responsabilités de l’investigateur
Investigateur : collection de tous les EI dans un cahier de
recueil des données du patient (CRF ou eCRF)
Causalité à Intensité (grade
1 réaction = 1 ligne
déterminer 1,2,3,4,5)
Evolution à
compléter
/!\ si intensité de grade 3,4 ou 5
= formulaire EIG à remplir et à envoyer au promoteur dans les 24h 32Responsabilités de l’investigateur
33Responsabilités de l’investigateur
Informations minimales devant figurer sur la fiche d’EIG
• Un investigateur
• Un patient
• Un médicament expérimental
• Un évènement indésirable*
* Si possible : sous forme de diagnostic
34Responsabilités de l’investigateur
Autres informations à faire figurer sur la fiche d’EIG
(dans la mesure du possible) :
En lien avec le patient :
• Facteurs de risques
• Pathologies en cours
• Médicaments associés
En lien avec la chronologie du
ou des EIG :
• Dates des traitements
(début/arrêt/modification)
• Dates précises de l’EIG
Date de survenue d’un EIG
= date des 1ers symptômes ≠ date d’hospitalisation
35Responsabilités de l’investigateur
Réaliser un descriptif complet de l’EIG
(dans la mesure du possible) :
• Description clinique et/ou
biologique
• Recherche d’autres causes
• Modalités de prise en
charge
• Evolution de l’évènement
Diagnostic médical précis = meilleur codage de l’EIG
36Responsabilités de l’investigateur
Evaluation du lien de causalité (imputabilité) :
L’investigateur évalue le lien de causalité entre
- d’une part l’EIG
- et d’autre part le ou les ME / la recherche
Bien penser à l’indiquer sur la fiche de déclaration d’EIG
37Responsabilités de l’investigateur
Recueil des EIG
= vigilance des
essais cliniques
Ne pas oublier de :
• Cocher un ou
plusieurs critères
de gravité
• Dater & signer
38Responsabilités de l’investigateur
…une fois la déclaration d’EIG envoyée :
• L’investigateur suit les évènements jusqu’à leur
résolution / stabilisation
• L’investigateur transmet au promoteur les données de
suivi
• L’investigateur fournit au promoteur toutes les
informations nécessaires à la bonne évaluation du cas
Une déclaration d’EIG bien remplie
= qualité et fiabilité des données de sécurité
= moins de queries pour les investigateurs et le promoteur = gain de temps
39Responsabilités de l’investigateur (9)
Suivi des EIG
Période de surveillance : définie dans le protocole
Début du Fin du
traitement traitement
Consentement Fin de l’essai
signé
Si EIG susceptible d’être en lien avec le médicament expérimental /
procédures de recherche
pas de limitation dans le temps
40Responsabilités de l’investigateur
Grossesse & allaitement
Si exposition fœtale ou durant l’allaitement à
un médicament expérimental/placebo
Même si absence d’EI
Eléments à rapporter au promoteur
Permet d’obtenir des informations importantes sur
l’issue de la grossesse / l’allaitement
41En pratique que change la loi Jardé?
Obligations de notification des EIG/EI par l’investigateur au
promoteur
Loi de santé publique
(ancienne réglementation)
Recherche en
Recherche soins courants +
biomédicale Recherche non
interventionnelle
EIG : sans délai
Pas d’exigence
EI : selon le protocole
42En pratique que change la loi Jardé?
Obligations de notification des EIG/EI par l’investigateur au
promoteur
Loi Jardé
Recherche
Recherche Recherche non
interventionnelle à
interventionnelle interventionnelle
risques minimes
EIG : notifiés dans le cadre du
soin au service de vigilance
EIG : sans délai EIG : NA
concerné (pharmacovigilance,
EI : selon le protocole EI : selon le protocole
matériovigilance,
biovigilance…)
Ordonnance n°2016-800 du 16 juin 2016
EI : selon le protocole 43Responsabilités de l’investigateur
EIG à ne pas notifier dans les 24h au promoteur
• Evolution naturelle et habituelle de la pathologie
– Hospitalisation programmée (suivi pathologie étudiée)
– Aggravation de la pathologie étudiée…
• Circonstances particulières
– Hospitalisation pour une pathologie préexistante
– Hospitalisation programmée avant la recherche
– Admission pour raisons sociales ou administratives…
EIG à recueillir dans le cahier d’observation
44Responsabilités de l’investigateur
Rôle des Attachés de Recherche Clinique
• Sensibilisation des investigateurs
(obligations réglementaires & importance
de leur respect )
• Amélioration de la qualité des données
renseignées sur les formulaires d’EIG
(données manquantes, inexactes, illisibles,
incomplètes…)
BPC paragraphe 5.18.4
45Responsabilités du promoteur
46Responsabilités du promoteur
Promoteur = responsable de la vigilance de son essai clinique
Option 1 : Option 2 :
Il en assure la gestion Il peut la déléguer :
CRPV, prestataire (CRO)…
47Responsabilités du promoteur
Gestion des évènements indésirables graves
Procédures
Registre
Promoteur
48Responsabilités du promoteur
Mise en place d’un registre des évènements indésirables graves
Procédures écrites assurant la qualité :
• Du recueil des données
• De la documentation des cas
• De leur évaluation / validation
• De leur déclaration
• De leur archivage
49Responsabilités du promoteur
Pour le codage MedDRA®
(Medical Dictionary for Regulatory Activities)
• Fin des années 90 par ICH
• Dictionnaire (anglais ++ / traductions disponibles)
• Harmonisation : uniformiser la terminologie médicale
• Plus de 70 000 termes
• Hiérarchisation des termes / Arborescence
– Niveau le plus haut = SOC (System Organ Class) ; 27 SOC
– Puis HLGT puis HLT puis PT puis LLT
• « MedDRA® term selection: Points to consider »
= recommandations (modalités de codage)
• Mises à jour régulièrement par MSSO
(Maintenance and Support Services Organization)
50Mars 2016 :
27th SOC
Product Issues
Exemple :
LLT Rash cutané érythémateux
PT Rash érythémateux
HLT Rash, éruptions et exanthèmes
HLGT Affections dermiques et
épidermiques
SOC Affections de la peau et du
tissu sous-cutanée
51Responsabilités du promoteur
Gestion des évènements indésirables graves
Qualification
Procédures Evaluation
Registre
Promoteur
52Responsabilités du promoteur
Qualification / évaluation :
• De la gravité de l’EI (qualification en EIG)
• Du lien de causalité avec le ME
• Du caractère attendu/inattendu (brochure investigateur)
• De la qualification en EIG inattendu (EIGI = SUSAR)
ou en EIG attendu (EIGA)
Avis investigateur Avis promoteur Déclaration ANSM/EMA
Lié au ME Non lié au ME OUI
Non lié au ME Lié au ME OUI
53Responsabilités du promoteur
De l’évènement indésirable (EI) à l’effet indésirable grave inattendu (EIGI)
EIGI Sans délai
Effet Annuelle
indésirable
grave Promoteur
Evènement
indésirable grave
Investigateur
Evènement indésirable
54En pratique que change la loi Jardé?
Obligations de notification des EIG/EI par le promoteur
(recherche portant sur un médicament)
Loi de santé publique (ancienne réglementation) Loi Jardé
Rercherche biomédicale Recherche en soins Recherche
Recherche
courants et Recherche interventionnelle à
interventionnelle
Phase Pilote non interventionnelle risques minimes
Evènement à déclarer EIG inattendus (SUSAR) EIG inattendus (SUSAR) EIG inattendus (SUSAR)
Initial : 7 j Initial : 48 h Initial : sans délai
Décès / MEJPV
Délais de Suivi : 8 j Initial : 48 h Suivi : 8 j
déclaration Initial : 15 j Initial : 7 j Initial : 15 j
Pharmacovigilance Pharmacovigilance
Autres EIG
Suivi : 8 j Initial : 7 j Suivi : 8 j
FNS Sous 15 j Sous 15 j Sans délai
Rapports annuels de sécurité 1 fois par an 1 fois par an
55En pratique que change la loi Jardé?
Obligations de notification des EIG/EI par le promoteur
(recherche portant sur un dispositif médical)
Loi de santé publique (ancienne réglementation) Loi Jardé
Rercherche Recherche en soins Recherche Recherche
biomédicale courants et Recherche interventionnelle à interventionnelle
Evènement à déclarer EIG inattendus (SUSAR) EIG inattendus (SUSAR)
Initial : 7 j Initial : sans délai
Décès / MEJPV
Délais de Suivi : 8 j Suivi : 8 j
déclaration Initial : 15 j Initial : 15 j
Matériovigilance Matériovigilance
Autres EIG
Suivi : 8 j Suivi : 15 j
FNS Sous 15 j Sans délai
Rapports annuels de sécurité 1 fois par an 1 fois par an
56Responsabilités du promoteur
Gestion des évènements indésirables graves
Qualification
Procédures Evaluation
Registre
Promoteur
Déclaration
SUSAR/FNS
57Responsabilités du promoteur
Déclaration des effets indésirables graves inattendus (EIGI ou SUSAR) :
• A l'ANSM à declarationsusars@ansm.sante.fr
• Au CPP concerné
(uniquement les SUSAR survenus en France dans l'essai
concerné)
• Faire une déclaration à Eudravigilance :
https://eudravigilance.ema.europa.eu/human/index.asp
Doivent être déclarés toutes suspicions d’EIGI/SUSAR:
- survenues en France et en dehors du territoire national, au cours de l’essai
concerné
- liées à la même substance active, survenues au cours d’un autre essai (mené
dans un autre Etat membre ou hors UE) promu par le même promoteur ou un
autre promoteur appartenant à la même société mère ou lié par un accord
58Responsabilités du promoteur
Gestion des faits nouveaux de sécurité (FNS) :
Déclaration SANS DELAI
• A l'ANSM : aec-essaiscliniques@ansm.sante.fr
• Au CPP concerné
• A l’ARS de tous les lieux de recherche concernés lorsque
la recherche est menée chez des volontaires sains
FNS = toute nouvelle donnée pouvant conduire à :
- une réévaluation du rapport BR de la recherche ou du médicament
expérimental
- des modifications dans l’utilisation de ce médicament, dans la conduite de
la recherche, ou des documents relatifs à la recherche
- ou à suspendre ou interrompre ou modifier le protocole de la recherche ou
des recherches similaires.
NB : possible qu’un FNS soit aussi un EIGI
Dans ce cas : double déclaration (en tant que FNS et en tant qu’EIGI)
59Responsabilités du promoteur
Gestion des faits nouveaux de sécurité (FNS) :
Essais portant sur la première administration ou utilisation
d’un ME chez des personnes qui ne présentent aucune
affection (= volontaire sain) :
Tout EIG est constitutif d’un FNS.
Le promoteur doit :
- suspendre l’administration ou l’utilisation du ME chez les
personnes participant à la recherche dans l’attente de
l’adoption de mesures définitives
- prendre des mesures de sécurité urgentes appropriées
- informer sans délai l’ANSM, le CPP et l’ARS du fait
nouveau et le cas échéant, des mesures prises
60L’exemple de Biotrial
Quid de l’essai clinique promu par les laboratoires BIAL
et réalisé par la société BIOTRIAL à Rennes ???
61L’exemple de Biotrial
Chronologie de l’évaluation et du déroulement de l’essai clinique (1)
• « Etude en double aveugle, randomisée, versus placebo, combinant une
étude de doses uniques croissantes, une étude de doses multiples
croissantes, une étude interaction repas, dans le but d'évaluer la sécurité
d'emploi, la tolérance, les profils pharmacocinétique et pharmacodynamique
du BIA 10-2474 chez des volontaires sains » (réf. ANSM : 150565A-31 -
EudraCT n° 2015-001799-24).
• Centre de recherche BIOTRIAL situé à Rennes, volontaires sains hommes et
femmes âgés de 18 à 55 ans.
• Objectifs de l’essai étaient d’évaluer :
- la sécurité d’emploi et la tolérance du BIA 10-2474 après administration de
doses orales uniques et multiples
- l’effet repas sur la pharmacocinétique (PK) du BIA 10-2474
- la pharmacocinétique (PK) et la pharmacodynamie (PD) du BIA 10-2474.
http://ansm.sante.fr/content/download/84953/1072475/version/3/file/Chrono+point+dinfo+27.01.16+presse.pdf consulté le 23/03/2016
62L’exemple de Biotrial
Chronologie de l’évaluation et du déroulement de l’essai clinique (2)
Déroulement de l’essai, survenue des EIG et gestion de la situation
Protocole : 3 étapes pour cet essai
• 1ère étape : administration de doses orales uniques croissantes
- 8 doses croissantes testées, chaque dose testée sur une cohorte de 8
volontaires du 9 juillet 2015 (0,25 mg)au 9 octobre 2015 (100 mg)
- Aucun EIG n’a été porté à la connaissance de l’ANSM au cours de cette 1ère
partie.
• 2ème étape : interaction avec la nourriture
- Cohorte de 12 volontaires : à 2 reprises, 2 doses de verum à 40 mg, soit à jeûn
(21 octobre 2015), soit après un petit déjeuner riche en matières grasses (10
novembre 2015)
63
http://ansm.sante.fr/content/download/84953/1072475/version/3/file/Chrono+point+dinfo+27.01.16+presse.pdf consulté le 23/03/2016L’exemple de Biotrial
Chronologie de l’évaluation et du déroulement de l’essai clinique (3)
Déroulement de l’essai, survenue des EIG et gestion de la situation
Protocole : 3 étapes pour cet essai
• 3ème étape : administration de doses orales multiples et croissantes
- Chaque volontaire : traitement 1 fois/j pendant 10 j
- 5 doses croissantes (2,5 mg, 5 mg, 10 mg, 20 mg puis 50 mg) testées
(chaque dose étant testée dans une cohorte de 8 volontaires)
- Dans chaque cohorte : 6 verum et 2 placebo
1ere cohorte (dose de 2,5 mg) : du 6 octobre au 15 octobre 2015
2ème cohorte (dose 5 mg) : du 28 octobre au 6 novembre 2015
3ème cohorte (dose 10 mg) : du 17 au 26 novembre 2015
4ème cohorte (dose 20 mg) : du 9 au 18 décembre 2015
Aucun EIG porté à la connaissance de l’ANSM depuis la cohorte 1 et jusqu’à la fin
de la cohorte 4
64
http://ansm.sante.fr/content/download/84953/1072475/version/3/file/Chrono+point+dinfo+27.01.16+presse.pdf consulté le 23/03/2016L’exemple de Biotrial
Chronologie de l’évaluation et du déroulement de l’essai clinique (4)
Déroulement de l’essai, survenue des EIG et gestion de la situation
Protocole : 3 étapes pour cet essai
• 3ème étape : administration de doses orales multiples et croissantes
5ème cohorte (dose 50 mg) : mardi 6 janvier 2016 : début du traitement
• Dimanche 10 janvier 2016 :
- J5 de 50 mg aux 8 volontaires de la cohorte 5
- date d’apparition des symptômes chez un des volontaires de la cohorte : hospitalisation le soir
de ce volontaire.
• Lundi 11 janvier 2016 :
- Les 7 autres volontaires de la cohorte 5 6ème dose de traitement le matin
- IRM chez le volontaire hospitalisé dont l’état clinique s’est aggravé (coma)
- Interruption de l’essai clinique en accord entre le promoteur BIAL et la société BIOTRIAL.
• Entre le mercredi 13 et le vendredi 15 janvier 2016 : hospitalisation des 5 autres volontaires
de la cohorte 5 traités par le verum.
• Jeudi 14 janvier 2016 :
65
L’ANSM est informée par BIOTRIAL de la survenue d’EIG dans le cadre de l’essai.L’exemple de Biotrial
Manquement majeurs dans la gestion de crise
• Nouvelle administration de la molécule chez les autres volontaires sans s’être tenu
suffisamment informé de l’état de santé du 1er volontaire hospitalisé
• L’AVC n’a d’abord pas été relié à l’étude :
- d’autres patients ont dû être impactés avant qu’un lien soit suspecté
- les autres volontaires sains n’ont pas été informé de cette donnée
- donc n’ont pas pu donner un consentement parfaitement éclairé
• aurait dû être considéré comme un Fait Nouveau de Sécurité et aurait dû être
déclaré sans délai à l’ANSM et non pas 4 jours après l’hospitalisation du 1er patient
66L’exemple de Biotrial
Et a posteriori…
• Constat (ANSM & IGAS) :
répartition des rôles et des responsabilités (déclaration FNS et EIGI) non clairement
définie dans les documents contractuels entre le prestataire et le promoteur
• Publication par le ministère des affaires sociales et de la santé de la circulaire
N°DGS/PP1/2016/61 du 1er mars 2016 relative aux déclarations des FNS et des EIG
survenant au cours des essais cliniques :
« S’agissant des essais sur le volontaire sain, pour tout effet indésirable grave conduisant à
une hospitalisation, il est demandé de le considérer comme un fait nouveau et de le
déclarer sans délai à l’ANSM et au CPP à compter du jour où le promoteur en a eu
connaissance.
Un tel évènement doit conduire à la suspension immédiate de l’essai jusqu’à la
démonstration de l’absence de danger pour les volontaires sains.
Par ailleurs, ceux-ci devront systématiquement être informés et leur consentement obtenu
avant toute nouvelle administration du produit à l’étude. »
• Communication de l’ANSM (31/03/2016) : mesures de sécurisation des essais cliniques
de phase I sur les volontaires sains - Point d‘information (circuit prioritaire au sein de
l’ANSM) :
https://ansm.sante.fr/S-informer/Points-d-information-Points-d-information/Mesures-de-securisation-des-
essais-cliniques-de-phase-I-sur-les-volontaires-sains-Point-d-Information 67Responsabilités du promoteur
Gestion des évènements indésirables graves
Qualification
Procédures Evaluation
Registre
Promoteur
Déclaration
Comité de SUSAR/FNS
surveillance
(CSI/DSMB)
68Responsabilités du promoteur
Mise en place d’un Comité de Surveillance Indépendant (CSI ou DSMB
(Data and Safety Monitoring Board)
= groupe d’experts indépendant (3 membres minimum ; nombre impair)
Essentiellement pour les EC en double aveugle mais pas seulement…
• Examen régulier des données de sécurité
recueillies
• Convocation/réunion si nécessaire et/ou à
fréquences prédéfinies (cf protocole)
• Emission de recommandations au promoteur
quant à la poursuite, modification, arrêt de
l’essai
69Responsabilités du promoteur
Gestion des évènements indésirables graves
Rapports de
sécurité
(RAS/DSUR)
Qualification
Procédures Evaluation
Registre
Promoteur
Déclaration
Comité de SUSAR/FNS
surveillance
(CSI/DSMB)
70Responsabilités du promoteur
Rapports de sécurité ou Development Safety Update Report (DSUR)
ICH guideline E2F
• 3 parties :
– Analyse de la sécurité des personnes
– Liste de tous les EIG (attendus et inattendus)
– Tableaux de synthèse de tous les EIG
• A quelle fréquence?
Une fois par an à la date anniversaire
= date de 1ère autorisation d’essai clinique
/!\ à rendre dans un délai de 60 jours calendaires
• A qui ?
– ANSM
– CPP
71Exemple : table des matières – DSUR
72Responsabilités du promoteur
Gestion des évènements indésirables graves
Rapports de
sécurité
(RAS/DSUR)
Qualification
Procédures Evaluation
Registre
Promoteur
Gestion
de l’insu
Déclaration
Comité de SUSAR/FNS
surveillance
(CSI/DSMB)
73Responsabilités du promoteur
Gestion de l’insu
ICH guideline E6
Bonnes pratiques cliniques (BPC)
• Procédure de décodage :
Documente comment, en cas d'urgence, l'identité du ME
en aveugle peut être révélée sans compromettre l’insu
pour le traitement des sujets restants
• Investigateur :
– respect levée d’insu tels qu’établit dans le protocole
– levée d’insu (cas individuels) que si la connaissance
de cette information modifie la prise en charge du
patient
– documentation et explication au promoteur de la
raison de la levée d’insu
74Responsabilités du promoteur
Gestion de l’insu et EIG inattendus (SUSAR)
• Levée d’insu par le promoteur avant déclaration de l’EIGI
• 3 situations possibles
– le produit administré = le produit expérimenté
suspicion d’EIGI = déclaration
– le produit administré = les comparateur
réévaluation et selon, déclaration ou non de l’EIG
– le produit administré = le placebo
pas de déclaration sauf si lié au placebo (ex: effet dû à
un excipient)
75En bref…
76Investigateur
Promoteur ANSM
Déclaration EIG Critères EVCTM
avec a minima réglementaires
les 4 critères pour déclaration CPP
aux AC? Investigateurs
DSMB
Safety
database
Responsabilité investigateur : Responsabilité promoteur :
déclaration sans délai déclaration sans délai ou
15j selon situations
77Investigateur
EIG Déclaration sans délai
Promoteur
Registre / qualification
EIG EIGA EIGI EMA (EVCTM)
Requalification
ANSM
CPP
Rapport annuel de sécurité
Investigateurs
DSMB
Délais de transmission
7879
MERCI POUR VOTRE ATTENTION
80Vous pouvez aussi lire

















































