Session plénière du GDR Ultrafast Phenomena 28 30 septembre 2016 - CEA
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Session plénière du GDR Ultrafast Phenomena
28‐30 septembre 2016
http://gdrupilm.univ‐lyon1.fr/News.html
Cité internationale universitaire de Paris
http://www.ciup.fr/salon‐honnorat/
Recueil des résumés des présentations orales et posters
1
Le GDR U.P. consacré aux phénomènes ultrarapides a été officiellement créé le 01 janvier
2016 pour une durée de 5 ans. Il rassemble la communauté française des expérimentateurs et
théoriciens sʹintéressant aux phénomènes aux échelles de temps ultrabrèves : attoseconde,
femtoseconde et picoseconde et intervenant dans tous les états de la matière (milieu dilué,
solide, nanométrique, liquide et plasma).
La première réunion du GDR du 28 au 30 septembre 2016 à la Cité Universitaire
Internationale de Paris est une occasion importante de rapprochement entre les équipes
françaises et une démonstration de l’émulation scientifique qui caractérise notre communauté.
Nous vous remercions d’y participer et souhaitons, avec votre aide, une grande réussite
scientifique au GDR U.P.
Bien cordialement,
Franck LÉPINE & Lionel POISSON
Le bureau du GDR Ultrafast Phenomena :
Pascale CHANGENET‐BARRET (LOB), Danielle DOWEK (ISMO), Valérie HALTE (IPCMS),
Lionel POISSON (LIDYL), Patrick MARTIN (CELIA), Phuong Mai DINH (LPT), Lamri ADOUI
(CIMAP), Vincent DEWAELE (LASIR), Jérôme FAURE (LOA) et Franck LÉPINE (ILM)
2
Programme
28 septembre 2016
12h00‐14h00 Accueil Café/Lunch box Accueil
14h00‐14h20 Franck Lépine
Ouverture et présentation
Lionel Poisson
du GdR Ultrafast Phenomena
Bureau du GDR
Modérateur : Pascal Ruello Session : Spin & Magnétisme
14h20‐14h40 C. Richter
LPMS Femtosecond spin dynamics by photoemission
(Cergy‐pontoise)
14h40‐15h00 V. Halté
Dynamique dʹaimantation induite par impulsions lasers fs
IPCMS
dans des nanoparticules magnétiques
(Strasbourg)
15h00‐15h20 J. Luning
Lʹapport des impulsions XUV femtosecondes à lʹétude de la
LCPMR
dynamique dʹaimantation ultrarapide
(Paris)
Modérateur : Philippe Martin Session : Optique ultrarapide
15h20‐15h35 T. Gustavsson
LIDYL Présentation du réseau FEMTO
(Saclay)
15h35‐15h40 P. Monot
Offre de temps laser du comité de programme
LIDYL
LOA‐IRAMIS CELIA
(Saclay)
15h40‐16h00 C. Spezzani
Status report of the OPT2X project: advanced equipment for
LPS
ultrafast XUV beamline technology in Paris‐Saclay
(Orsay)
16h00‐16h30 Pause café
Modérateur : Agathe Espagne Session : Dichroïsme circulaire
16h30‐16h50 Y. Mairesse
Dynamiques ultrarapides de molécules chirales sondées par
CELIA
dichroisme circulaire de photoélectrons
(Bordeaux)
16h50‐17h10 F. Hache
Dynamique des G‐quadruplexes dʹADN étudiée par
LOB
dichroïsme circulaire résolu en temps
(Palaiseau)
Modérateur : Valérie Véniard Session : Théorie
17h10‐17h30 P‐A. Hervieux
HHG by nonlinear resonant excitation of surface plasmon
IPCMS
modes in metallic nano‐particles
(Strasbourg)
17h30‐17h50 A. de la Lande
Compétition entre Migration de Charge et Transfert de
LCP
Proton Ultrarapide Impliquant l’Espèce H2O°+
(Paris)
17h50‐20h30 Réunion du Bureau
3
29 septembre 2016
09h00‐09h30 Accueil café
Modérateur : Dominique Vernhet Session : Dynamique d’ionisation
09h30‐09h50 P. Salières
LIDYL Dynamique attoseconde de l’auto‐ionisation
(Saclay)
09h50‐10h10 V. Loriot
Dynamique attoseconde et femtoseconde dans les HAPs par
ILM
imagerie de vitesse
(Lyon)
10h10‐10h30 J. Rangama Fragmentation de dimères de gaz rares : Cartographie de la
CIMAP collision et processus de relaxation
(Caen)
10h30‐11h00 Pause café
Modérateur : Niloufar shafizadeh Session : Photo‐réactivité en phase diluée
11h00‐11h20 A. Czasch
Single particle counting detectors and ultra‐fast phenomena
Roentdek
11h20‐11h40 A. Röder
Dynamique ultrarapide du radicale benzyle en phase
LIDYL
gazeuse
(Saclay)
11h40‐12h00 E. Suraud
LPT Clusters and molecules in extreme light
(Toulouse)
12h00‐12h20 F. Gatti
Quantum simulations with laser pulses for tomorrow’s
ICGM
chemistry
(Montpellier)
12h20‐14h00 Buffet
Modérateur : Laura Antonucci Session : Sources laser haute cadence
14h00‐14h20 E. Cormier
Laser intense à très haute cadence pour l’étude des
CELIA
phénomènes ultrarapides
(Bordeaux)
14h20‐14h40 N. Forget
Source infrarouge à haute cadence pour la génération X‐UV
FastLite
14h40‐15h00 M. Hanna
Source paramétrique ultrarapide dans lʹinfra‐rouge moyen à
LCF
taux de répétition élevé
(Palaiseau)
15h00‐15h30 Pause café
Modérateur : Pascale Changenet‐Barret Session : Dynamique moléculaire en phase liquide
15h30‐15h50 P. Plaza
Etudes par spectroscopie ultrarapide de la réactivité
PASTEUR‐ENS
photoinduite de protéines photoactives
(Paris)
15h50‐16h10 M. Joffre
Spectroscopie multidimensionnelle infrarouge dans
LOB
l’hémoglobine
(Palaiseau)
16h10‐16h30 S. Aloïse
The photochemistry of inverse dithienylethene switches
LASIR
understood
(Lille)
16h30‐19h30 Session poster / Cocktail
4
30 septembre 2016
09h00‐09h30 Accueil café
Modérateur : Adeline Bonvalet Session : Cohérence vibrationnelle/rotationnelle
09h30‐09h50 E. Hertz
Alignement moléculaire par impulsions laser ultra‐brèves :
ICB
production, mesure et applications récentes
(Dijon)
09h50‐10h10 J. Leonard
IPCMS Spectroscopie de Cohérences vibrationnelles en phase liquide
(Strasbourg)
10h10‐10h30 A. Crut
Time‐domain investigations of the vibrational and cooling
ILM
dynamics of metal and hybrid nano‐objects
(Lyon)
10h30‐11h00 Pause café
Modérateur : Patrick Martin Session : Phase condensée
11h00‐11h20 M. Lorenc
From molecular switching to material transformation:
IPR
revisiting the spin crossover
(Rennes)
11h20‐11h40 C. Hynes
PASTEUR‐ENS Dynamical Disorder in the DNA Hydration Shell
(Paris)
11h40‐12h00 E. Papalazarou
LPS Out‐of‐equilibrium electron dynamics in topological matter
(Orsay)
12h00‐12h20 B. Chalopin Champ fort au bout d’une pointe: nouveaux outils pour la
LCAR photoémission et le contrôle cohérent
(Toulouse)
12h20‐14h00 Buffet
Modérateur : Annie Klisnick Session : Plasma & Haute intensité
14h00‐14h20 F. Quéré
Interaction d’impulsions laser ultra‐intenses avec des plasmas
LIDYL
denses : différents mécanismes d’accélération d’électrons
(Palaiseau)
14h20‐14h40 A. Ricci SourceLAB : From ultrafast laser to innovative targetry and
SourceLab new sources of light and particles
14h40‐15h00 F. Courvoisier
Mise en forme spatiale dʹimpulsions femtosecondes:
Femto‐ST
Filamentation et micro‐explosions dans les solides
(Besançon)
15h00‐15h20 J. Faure
LOA Sources d’électrons ultrabrèves pour la diffraction d’électrons
(Palaiseau)
15h20‐15h30 Cloture
5
Connectez‐vous sur le réseau (Login to the
network): CIUP_Colloques
Nom d’utilisateur (User Name) tapez:
0916honnorat
Mot de passe (Password) tapez: 0916honnorat
Merci de ne pas afficher les codes en
dehors des salons. (Thank you not to post
codes outside rooms)
6
Présentations Orales
Femtosecond spin dynamics by photoemission
C. Cacho1, M. Battiato2, A. Crepaldi3, J. Braun4, F. Cilento3, M. Zacchigna3, M. C. Richter6, O.
Heckmann6, E. Springate1, S. S. Dhesi8, M. Grioni9, J. Minar4, F. Parmigiani5, J.‐M. Mariot10, K.
Hricovini6
1 Central Laser Facility, STFC Rutherford Appleton Laboratory, Harwell, United Kingdom
2 Institute of Solid State Physics, Vienna University of Technology, Wien, Austria
3 Elettra ‐ Sincrotrone Trieste, Basovizza, Trieste, Italy
4 LMU Munich, Chemistry Department, Germany
5 C.N.R. ‐ I.O.M., Trieste, Italy
6 LPMS, Université de Cergy‐Pontoise, France
8 Diamond Light Source, Chilton, Didcot, Oxfordshire, United Kingdom
9 Institute of Condensed Matter Physics (ICMP), Ecole Polytechnique Fédérale de Lausanne (EPFL),
Lausanne, Switzerland
10 Sorbonne Universités, UPMC Université Paris 06, CNRS, LCP–MR, France
We will present studies implying the use of time‐ and spin‐resolved APRES measurements with
samples of two groups of materials of great interest in todayʹs material research: the topological
insulator Bi2Se3 and the half‐metal Fe3O4, both of application potential for spintronic devices.
The samples are optically pumped with infrared photons (fundamental) and probed with the
third or fourth harmonic (4.5 ‐ 6 eV) femtosecond laser radiation in order to explore the
electronic relaxation above the Fermi level in both spin channels. The ARPES measurement is
performed with a spin‐resolved electron Time‐of‐Flight analyser which allows to resolve the
spin of the photoelectrons over a few orders of magnitude on the photoemission signal.
Working in close collaboration with theorists we were able to extract fundamental knowledge
in both cases.
7
Dynamique dʹaimantation induite par impulsions lasers femtosecondes dans des
nanoparticules magnétiques
V. Halté
IPCMS Strasbourg
Nous comparons, dans ce travail, la dynamique d’aimantation d’assemblées de
nanoparticules de Fe3O4 (magnétite) et de ‐Fe203 (maghémite) issues du même bain et oxydées
de manière contrôlée par recuit thermique. Alors que la désaimantation ultrarapide s’opère
après la thermalisation des électrons dans les nanoparticules de magnétite, les deux processus
sont simultanés après le recuit thermique qui les transforme en maghémite. Nous pensons que
cette accélération de la désaimantation dans la phase oxydée de la magnétite est due à un
renforcement du couplage super‐échange anti‐ferromagnétique qui régit le magnétisme dans
ces deux systèmes. Nous montrons ainsi le rôle majeur joué par l’interaction d’échange dans la
désaimantation ultrarapide qui suit l’excitation d’un matériau magnétique par une impulsion
laser sub‐picoseconde.
8
Lʹapport des impulsions XUV femtosecondes à lʹétude de la dynamique dʹaimantation
ultrarapide
J. Luning
LCPMR Paris
9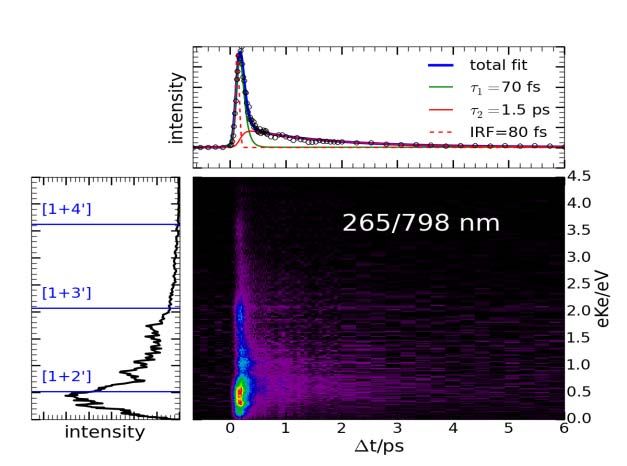
Présentation du réseau FEMTO
T. Gustavsson
LIDYL Saclay
Offre de temps laser du comité de programme
LOA‐IRAMIS CELIA
P. Monot
LIDYL Saclay
10Status report of the OPT2X project: advanced equipment for ultrafast
XUV beamline technology in Paris‐Saclay
C. Spezzani (LPS ORSAY) on behalf of the Opt2X consortium:
Danielle Dowek, Charles Bourassin‐Bouchet, Bertrand Carré, Maël Delhinger, Franck Delmotte,
David Dennetiere, Sébastien Derossi, David Garzella, Stefan Haessler, Sophie Kazamias, Annie
Klisnick, Julien Lenfant, Rodrigo Lopez‐Martens, Marino Marsi, Jean‐Michel Mestdagh, Laurent
Nahon, Lionel Poisson, François Polack, Thierry Ruchon, Philippe Zeitoun
The OPT2X project in one of the LIDEX projects of the University of Paris‐Saclay which aims at
providing the Paris Saclay scientific community with advanced experimental instrumentation
for the optimal exploitation of ultrafast laser‐based light sources operating in the XUV range. It
joins the effort of nine different laboratories of the Paris‐Saclay campus, merging an
interdisciplinary expertise that includes high‐level competences on ultra‐short laser‐based light
sources development, ultrafast science applications on solid state, gas phase and plasma
physics, advanced optics and optical methods, synchrotron radiation based technology.
I will report on the recent achievements obtained in the framework of the OPT2X project. In
particular, I will focus on the development of the two XUV beamlines for the ATTOLAB‐FAB10
and LASERIX light sources of the Paris‐Saclay campus.
11Dynamiques ultrarapides de molécules chirales sondées par dichroisme
circulaire de photoélectrons
Y. Mairesse
CELIA, Université Bordeaux‐CNRS‐CEA
Les molécules chirales existent sous deux formes, nommées énantiomères, qui sont images lʹune
de lʹautre dans un miroir mais non superposables. Deux énantiomères ne peuvent être
distingués quʹà travers leur interaction avec un autre objet chiral. Les processus chimiques
énantiospécifiques jouent un rôle crucial en biologie car de nombreux constituants de base du
vivant sont chiraux.
La compréhension des mécanismes en jeu dans la réactivité chirale requiert de mesurer les
dynamiques de molécules chirales à leurs échelles naturelles, dans le domaine femtoseconde
voire attoseconde. Les impulsions de lumière polarisées circulairement peuvent être
employées pour distinguer deux énantiomères, via des processus chiroptiques.
Cependant, la grande majorité de ces processus est interdite dans lʹapproximation dipolaire
électrique, et ces effets sont donc très faibles. Leur utilisation pour lʹétude de molécules en
phase gaseuse est donc très difficile. Le dichroïsme circulaire de photoélectrons (DCPE) est
une exception à cette règle: cʹest un effet purement dipolaire électrique, et très intense.
Le DCPE émerge lorsquʹun rayonnement polarisé circulairement ionise des molécules chirales.
La distribution angulaire des photoélectrons émis présente une forte asymmétrie selon lʹaxe de
propagation de la lumière. Nous verrons que cet effet peut être observé quel que soit le régime
dʹionisation: un photon, multiphon, ou encore ionisation tunnel. Nous présenterons alors les
résultats dʹune expérience pompe‐sonde dans laquelle nous avons suivi la relaxation dʹun
paquet dʹondes de Rydberg dans une molécule chirale en mesurant le DCPE dépendant du
temps. Le DCPE révèle des dynamiques invisibles dans le spectre de photoélectrons intégré
angulairement, ce qui confirme son intérêt et sa grande sensibilité comme sonde ultrarapide.
12Dynamics of DNA G‐quadruplex structures by time‐resolved circular dichroism
Marco Schmid, Pascale Changenet‐Barret, and François Hache
Laboratoire dʹOptique et Biosciences,Ecole Polytechnique, CNRS, INSERM ; 91128 Palaiseau cedex,
France
G‐quadruplexes are DNA structures composed of stacked guanine quartets which are
of growing interest due to their prevalence in many important regions, such as telomeres and
regulatory regions of many oncogene promoters [1]. Manipulation and stabilization of these
DNA structures by specific ligands establish a new and promising field of cancer therapeutics.
In this paper, we investigate the folding/unfolding equilibrium of the quadruplex Tel21
(TTA(GGGTTA)3) by time‐resolved absorption and circular dichroism measurements coupled
with a T‐jump experiment [2]. Circular dichroism spectroscopy is performed thanks to an
original technique where a unique pulse serves as probe for the absorption of right and left‐
polarized light, yielding self‐referenced signals.
Directly after heating by a nanosecond IR laser pulse, we observe a decrease in
absorption and circular dichroism which is assigned to quadruplex unfolding. This is followed
by an increase reflecting the refolding dynamics once the temperature jump is over.
Investigations on dynamics for larger time scales are expected to reveal more information and
confirm further our results.
Abs Change (x 100), CD (L x 1000)
0.0
-0.1
-0.2
-0.3
-0.4
-0.5
-0.6
-0.7
0 20 40 60 80 100 120 140 160 180 200 220
Time (ms)
Figure 1 : Change in absorption and circular dichroism of Tel21 at 293 nm following a 5°C
temperature jump.
[1] J. Huppert, Phil. Trans. R. Soc. A 365, 2969 (2007).
[2] L. Mendonça, A. Steinbacher, R. Bouganne, F. Hache, J. Phys. Chem. B 118, 5350 (201
13High‐harmonic generation by nonlinear resonant excitation of surface plasmon modes in
metallic nanoparticles
P.A. Hervieux
IPCMS Strasbourg
The nonlinear electron dynamics in metallic nanoparticles is studied using a hydrodynamic
model that incorporates most quantum many‐body features, including spill‐out and nonlocal
effects as well as electron exchange and correlations. We show that, by irradiating the
nanoparticle with a chirped laser pulse of modest intensity (autoresonance), it is possible to
drive the electron dynamics far into the nonlinear regime, leading to enhanced energy
absorption and complete ionization of the nanoparticle on a time scale of the order of 100 fs. The
accompanying radiated power spectrum is rich in high‐order harmonics.
14Compétition entre Migration de Charge et Transfert de Proton Ultrarapide Impliquant
l’Espèce H2O°+
Karwan Ali Omar, Aurélien de la Lande
Laboratoire de Chimie Physique, Université Paris‐Sud, CNRS, Université Paris Saclay
Le cation radical H2O°+ est la première espèce produite lors de lʹinteraction entre rayons
ionisants et milieux aqueux. Cette espèce a une durée de vie extrêmement courte allant de
quelques dizaines à quelques centaines de femtosecondes selon les études. La voie réactive
communément admise est celle de la perte dʹun proton pour donner le radical hydroxyle HO°,
le proton étant quant à lui capté par une molécule dʹeau pour donner le cation hydronium H3O+.
Dans cette présentation je présenterai des résultats de simulations numériques de la dynamique
de lʹeau ionisée au voisinage dʹune molécule de benzène. La démarche suivie repose dʹune part
sur la réalisation de simulations de dynamique électronique attoseconde par des techniques de
TDDFT (Time‐Dependent Density Functional Theory), et dʹautre part sur la réalisation de
dynamique moléculaire de type Born‐Oppenheimer. Cette étude a pour objectif dʹétudier la
compétition entre le transfert de proton susmentionné et la réaction dʹoxydation du benzène par
H2O°+. Nous montrons que lʹétape de transfert dʹélectron est intimement liée à lʹétat de
délocalisation de la lacune électronique.
15Dynamique attoseconde de l’auto‐ionisation
V. Gruson1, L. Barreau1, À. Jiménez‐Galan2, F. Risoud4, J. Caillat4, A. Maquet4, B. Carré1, F.
Lepetit1, J‐F. Hergott1, T. Ruchon1, L. Argenti2, R. Taïeb4, F. Martin2,3, P. Salières1*
1 LIDYL, CEA, CNRS, Université Paris‐Saclay, CEA Saclay, 91191 Gif‐Sur‐Yvette, France.
2 Departamento de Quimica, Modulo 13, Universidad Autonoma de Madrid, 28049 Madrid, Spain.
3 Instituto Madrileño de Estudios Avanzados en Nanociencia, Cantoblanco, 28049 Madrid, Spain.
4 Sorbonne Université, UPMC Université Paris 6, UMR 7614, LCPMR, 11 rue Pierre et Marie Curie 75231
Paris Cedex 05, France and CNRS, UMR 7614, LCPMR, Paris, France.
L’amplitude et la phase des paquets d’ondes sont les informations nécessaires pour accéder à
la dynamique des systèmes quantiques. Cependant, une caractérisation directe dans le domaine
temporel est hors d’atteinte pour les dynamiques électroniques ultrarapides, en particulier
celles reliées à la photoionization résonnante. Les développements récents de la spectroscopie
attoseconde sur les plans théorique [1, 2] et expérimental [3] ont ouvert la possibilité d’étudier
ces dynamiques. Nous montrons que l’interférométrie électronique résolue spectralement permet la
mesure complète de l’amplitude et de la phase d’un paquet d’ondes électronique créé au
voisinage d’une résonance d’auto‐ionisation dans l’hélium [4]. Des répliques du paquet
résonnant, obtenues par transition à deux photons, interfèrent avec des paquets d’onde de
référence formés dans le continuum ‘plat’, permettant la reconstruction temporelle directe du
paquet résonnant émis dans le continuum. Ceci permet ensuite d’accéder à la façon dont
la résonance d’auto‐ionisation se construit à l’échelle attoseconde par une analyse temps‐
/
fréquence basée sur la Transformée de Fourier limitée [1]: ,
(1) montrant comment le spectre se construit jusqu’au temps d’accumulation . Nos résultats,
en excellent accord avec les simulations, ouvrent la perspective à la fois d’études détaillées de la
dynamique de photoémission gouvernée par les corrélations électroniques, et le contrôle
cohérent de paquets d’ondes électroniques ‘structurés’.
Figure 1. Reconstruction expérimentale de l’évolution temporelle
de la résonance de Fano de l’hélium 2s2p à l’aide de
l’interférométrie électronique résolue spectralement. Le spectre
d’énergie des photoélectrons est tracé en fonction du temps
d’accumulation utilisé pour la Transformée de Fourier inverse.
L’évolution du spectre est tracée pour variant de ‐10 fs à 20 fs
avec ∆ = 1 fs. La chronologie de la formation de la résonance
peut être interprétée dans le formalisme de Fano : jusqu’à ~3 fs
(bleu), un spectre Gaussien émerge, reflétant la forme spectrale du
rayonnement attoseconde utilisé pour ioniser : la transition directe
vers le continuum domine. Ensuite, le chemin résonnant
commence à contribuer lorsque l’état doublement excité 2s2p se
désexcite dans le continuum (durée de vie : 17 fs): des interférences
spectrales se construisent jusqu’à ~20 fs, convergeant vers un profil
de raie asymétrique caractéristique du profil de Fano.
[1] Wickenhauser M et al 2005 Phys. Rev. Lett. 94, 023002
[2] Jiménez‐Galán A et al 2014 Phys. Rev. Lett. 113, 263001
[3] Kotur M et al 2016 Nat. Commun. 7, 10566
[4] Gruson V, Barreau L et al 2016 Science, in press
16Dynamiques attosecondes et femtosecondes dans les PAHs
V. Loriot, A. Marciniak, G. Karras, C. Bordas, F. Catoire, L. Quintard, E. Constant et F. Lépine
ILM, Institut Lumière Matière, Univ. Lyon, Université Claude Bernard Lyon 1, CNRS, F‐69622,
VILLEURBANNE, France
Nous présenterons la ligne de lumière de l’ILM permettant de résoudre temporellement
des dynamiques de molécules complexes (telles que les PAH) induites par un train
d’impulsions UVX attosecondes et sondées par une impulsion courte infrarouge. Plusieurs
mécanismes ont été isolés à travers la mesure de différentes observables comme le rendement
d’ions ou la distribution de vitesses des photoélectrons résolus aux échelles de temps
femtosecondes et attosecondes. Cette présentation regroupera les dernières avancées obtenues
sur les mécanismes de relaxation non‐adiabatique des PAH photoexcités dans l’UVX ainsi que
des mesures de phases de diffusion à l’échelle attoseconde. Ce type dʹapproche ouvre de
nouvelles perspectives à travers l’utilisation des impulsions ultracourtes UVX dans le domaine
de la physique moléculaire attoseconde.
17Fragmentation de dimères de gaz rares : Cartographie de la collision et processus de
relaxation
J. Rangama
CIMAP Caen
Single particle counting detectors and ultra‐fast phenomena
A. Czasch
Roentdek
18Dynamique des radicaux isolés en échelle femtoseconde
Anja Röder1, Lionel Poisson2, Ingo Fischer1, Roland Mitric1, Alexander Humeniuk1
1
University of Wuerzburg, Am Hubland Süd 97074 Wuerzburg, Germany
2
CEA, Laboratoire Interactions, Dynamiques et Lasers-Bât 522 91191 Gif-sur-Yvette, France
anja.roeder@cea.fr
Les radicaux sont des intermédiaires importantes dans la combustion [1] et ils sont également impliqués dans la
formation des hydrocarbures aromatiques polycycliques(HAP), qui sont des précurseurs de suie. [2] Vu qu’ils sont
thermodynamiquement stables et cinétiquement instable, ils jouent un rôle important dans la formation des
molécules complexes dans l’espace et dans les nuages galactiques. [3]
Les radicaux sont produits à partir du précurseur nitrite via pyrolyse et ils sont ensuite examinés en phase gazeuse
en faisant des expériences pompe-sonde résolue en échelle femtoseconde. Utilisant la spectroscopie de masse et
la spectroscopie de photoélectrons permet d’examiner la durée de vie des états excités de ces radicaux.
Figure 1: Production du radical benzyle via pyrolyse
Le radical benzyle a été excite dans l’état D9 par un laser fs à 265 nm et ionisé ensuite soit par un laser de sonde à
798 nm ou à 398 nm. Les spectres de masses obtenu avec la sonde à 798 nm montrent deux temps de déclin (τ1=
80 fs; τ2=1.5 ps), contrairement aux spectres de masse obtenu avec une sonde de 398 nm, qui montrent qu’un
temps de déclin (τ1= 80 fs).
Les spectres de photoélectrons obtenu avec une sonde de 798 nm montrent, autre que les mêmes temps de
déclins que les spectres de masse, un spectre d’empreinte de Rydberg. Ce spectre correspond à une série P.
Avec 398 nm comme sonde on observe qu’un seul pic avec une dépendance temporelle.
Ces résultats peuvent être expliques en se basant sur des calculs théoriques effectués par la groupe de R. Mitric
(Würzburg). Ils postulent une dépopulation rapide du D9 qui correspond au déphasage du paquet d’onde, qui
ensuite se transfère plus lentement dans les état D1/D2. Vu que les spectres obtenus avec le laser 798 nm comme
sonde sont des spectres issus d’une ionisation multiphotonique, l’énergie totale déposée est supérieure à celle
d’une sonde à 398 nm, ce qui permet de d’observer le déclin d’états plus profonds.
.
Figure 2: Spectres de photoélectrons avec un laser de sonde à 798 nm (gauche) et à 398 nm (droit)
[1] Miller, J.A.; Kee, R.J.; Westbrook, C.K. Annu. Rev. Phys. Chem 1990, 41, 345
[2] Kohse-Höinghaus K.; Atakan B., Kasper T. Phys. Chem. Chem. Phys. 2002 4 2056-2062
[3] Herbst E. Chem. Soc. Rev. 2001, 30, 168
19Clusters and molecules in extreme light
P. M. Dinh1, P. G. Reinhard2, E. Suraud 1;3
1Laboratoire de Physique Théorique, UMR 5152, Université Paul Sabatier, 118 route de Narbonne,
F‐31062 Toulouse Cedex, France
2Institut für Theoretische Physik, Universität Erlangen Staudtstr. 7, D‐91058 Erlangen, Germany
3Physics Department, SUNY, University at Buffalo, Buffalo NY, USA
The progress in laser technology over the last decades has opened up new avenues for the
exploration of properties of clusters and molecules. A laser pulse is characterized by its
frequency but also by the laser intensity as well as the laser time profile. While for years the
variations of these parameters were heavily constrained by technology, the last two decades
and even more so the last years have seen tremendous increases in the range of attainable
parameters. This is true for intensity, which since the 1990ʹs can reach huge values which can
lead to very large energy deposits and possibly violent disintegration of the irradiated species.
But this is also true for the tuning of the time profile which can now be tailored up to time
scales of the order of magnitude of electronic motion and even below. This allows the follow up
of the detail of electronic dynamics at its own ʺnaturalʺ time. The latest breaktroughs were
attained in terms of laser frequency with the ongoing possibility of reaching very large
frequencies up the X domain. This opens up new possibilities of imaging which are
progressively being explored.
We shall discuss some of these directions of investigation, taking examples in cluster and
molecular physics. We shall especially discuss the case of high intensity and short time pulses
for which a sizable amount of results have already been attained. We shall also discuss in detail
the case of very short times (attoseconds) which are becoming more and more studied.
[1] Mechanisms of cluster ionization in strong laser pulses, U. Saalmann, C. Siedschlag,, J. M. Rost, J. Phys. B 39
(2006) R39.
[2] Non linear electron dynamics in metal clusters, F. Calvayrac, P. G. Reinhard, E. Suraud, C. Ullrich, Phys.
Reports 337(2000)493‐578
[3] Laser‐driven nonlinear cluster dynamics, Th. Fennel, K.‐H. Meiwes‐Broer, J. Tiggesbumker, P.‐G. Reinhard, P.
M. Dinh, E. Suraud, Rev. Mod. Phys. 82 (2010) 1793
[4] Probing Time‐Dependent Molecular Dipoles on the Attosecond Time Scale, C. Neidel et al Phys.Rev. Lett 111
(2013) 033001.
20Quantum simulations with laser pulses for tomorrow’s chemistry
F. Gatti
CTMM Institut Charles Gerhardt UMR‐CNRS 5253 University of Montpellier, France.
With the advent of femtochemistry and attospectrocopy [1,2] and the possibility to align
molecules with laser pulses, it becomes possible to measure and control the most subtle
quantum effects in molecules [3]. This opens the door for very important technological
applications on the long term, a ”tomorrow’s chemistry” working at the most elementary level
of chemical processes and based on quantum superposition [4].
We present several simulations with the Multi‐Configuration Time‐Dependent Hartree
(MCTDH) approach [5,6]. MCTDH is a general algorithm to solve the time‐dependent
Schrödinger equation for multidimensional dynamical systems consisting of distinguishable
particles. MCTDH can thus determine the quantal motion of the nuclei of a molecular system
evolving on one or several coupled electronic potential energy surfaces.
We present applications of control of elementary chemical processes with laser pulses for the
coupled motion of electrons and nuclei.
[1] M. F. Kling, C. Siedschlag, A. J. Verhoef, J. I. Khan, M. Schultze, T. Uphues, Y. Ni, M. Uiberacker, M. Drescher,
F. Krausz, and M. J. J. Vrakking. Control of electron localization in molecular dissociation. Science 312 (2006), 246.
[2] P. B. Corkum and F. Krausz. Attosecond science. Nat. Photon. 3 (2007), 381.
[3] C. B. Madsen, L. B. Madsen, S. S. Viftrup, M. P. Johansson, T. B. Poulsen, L. Holmegaard, V. Kumarappan, K.A.
Jorgensen, and H. Stapelfeldt. Manipulating the torsion of molecules by strong laser pulses. Phys. Rev. Lett. 102
(2009), 073007.
[4] ”Molecular Quantum Dynamics. From Theory to Applications” in Physical Chemistry in Action Ed. Springer
Verlag 2014. Editor : F. Gatti.
[5] H.‐D. Meyer, F. Gatti, and G. Worth, Multidimensional Quantum Dynamics: MCTDH Theory and Applications
Wiley‐VCH, 2009.
[6] See http://www.pci.uni‐heidelberg.de/tc/usr/mctdh/
21Laser intense à très haute cadence pour l’étude des phénomènes ultrarapides
E. Cormier
CELIA Bordeaux
Source infrarouge à haute cadence pour la génération X‐UV
N. Forget
FastLite
Source paramétrique ultrarapide dans lʹinfra‐rouge moyen à taux de répétition élevé
M. Hanna
LCF Palaiseau
22Etudes par spectroscopie ultrarapide de la réactivité photoinduite de protéines photoactives
Pascal Plaza,1,2 Ryan Martin,1,2 Agathe Espagne,1,2 Nadia Dozova,1,2 Dheerendra Yadav,1,2 Junpei
Yamamoto,3 Pavel Müller,4 Klaus Brettel,4 Fabrice Rappaport5
1 Ecole normale supérieure, PSL Research University, UPMC Univ Paris 06, CNRS, Département de
Chimie, PASTEUR, 24, rue Lhomond, 75005 Paris, France
2 Sorbonne Universités, UPMC Univ Paris 06, ENS, CNRS, PASTEUR, 75005 Paris, France
3 Graduate School of Engineering Science, Osaka University, 1‐3 Machikaneyama, Toyonaka, Osaka 560‐
8531, Japan
4 Institut de Biologie Intégrative de la Cellule (I2BC), CEA, CNRS, Univ Paris‐Sud, Université Paris‐
Saclay, 91198 Gif‐sur‐Yvette cedex, France
5 UMR7141, Institut de Biologie Physico‐Chimique, 13, rue Pierre et Marie Curie, 75005 Paris, France
Pour illustrer lʹactivité de recherche de lʹéquipe de photochimie ultrarapide au département
de chimie de lʹENS, on rapportera des travaux récents concernant lʹélucidation des mécanismes
primaires de photoactivation de deux protéines photoactives, une photolyase de type (6‐4) et et
la protéine fluorescente photocommutable Dronpa. La technique dʹinvestigation principalement
employée est la spectroscopie dʹabsorption transitoire en bande large, dans la région optique,
avec une résolution temporelle de lʹordre de la centaine de femtosecondes.
Les lésions de lʹADN induites par le rayonnement UV (principalement dimères cyclobutane de
pyrimidines et photoproduits dits (6‐4)) peuvent dans beaucoup dʹorganismes être
photoréparées par des flavoenzymes nommées photolyases. Pour que la photoréparation puisse
se produire, il est indispensable que le cofacteur FAD (flavine
adénine dinucléotide) soit sous sa forme entièrement réduite
(FADH–). Si cela nʹest pas le cas, une réaction photoinduite
secondaire et indépendante de la photoréparation, appelée
photoactivation, est capable de réduire la flavine oxydée dans
sa forme catalytiquement active. Nous avons spécifiquement étudié cette photoréduction pour
la photolyase (6‐4) de X. laevis. Le mécanisme réactionnel putatif est un transfert dʹélectron en
série le long dʹune chaîne de résidus tryptophane. Un effort particulier a été consacré à la
distinction spectroscopique entre espèces identiques (radicaux tryptophanyle) mais
différemment orientées, grâce à des mesures dʹanisotropie dʹabsorption transitoire.
Dronpa est une protéine fluorescente photochrome, structurellement analogue à la GFP mais
susceptible dʹêtre photocommutée de nombreuses fois entre un
fluorescent (ON) et un état non‐fluorescent (OFF). Les protéines de
ce type sont notamment très prometteuses en tant que sondes pour
la microscopie de super‐résolution (PALM...). Des structures
cristallographiques ont montré que la photocommutation de Dronpa
repose sur une isomérisation cis‐trans du chromophore,
accompagnée dʹune protonation/déprotonation de celui‐ci. Nous
avons étudié le mécanisme et la dynamique de la photoactivation de Dronpa, cʹest‐à‐dire de la
23photoconversion de OFF (trans protoné) à ON (cis déprotoné), sur une échelle de temps allant
de la centaine de femtosecondes aux millisecondes. Après le déclin de lʹétat excité dans le
régime picoseconde, nous avons observé une espèce transitoire portant la signature dʹun
isomère cis du chromophore à lʹétat fondamental, non encore déprotoné. Nous avons ensuite
montré que la déprotonation finale du chromophore, aboutissant à la formation de lʹétat ON, se
produit dans le régime microseconde.
24Spectroscopie multidimensionnelle infrarouge dans l’hémoglobine
Vincent Kemlin(1) , Adeline Bonvalet(1) , Cyril Falvo(2) , Christoph Meier(3) , Louis Daniault(1) ,
Thibault Vieille(1) , Jean‐Christophe Lambry(1) , Marten H. Vos(1) , Manuel Joffre(1)
(1) LOB, Ecole Polytechnique, CNRS, INSERM, Univ. Paris‐Saclay, 91128 Palaiseau, France
(2) ISMO, Univ. Paris‐Sud, CNRS, Univ. Paris‐Saclay, 91405 Orsay, France
(3) LCAR, IRSAMC, Univ. Paul Sabatier, CNRS, 31062 Toulouse, France
La spectroscopie multidimensionnelle dans les domaines de l’infrarouge et du visible constitue
une transposition au domaine optique de la Résonance Magnétique Nucléaire (RMN) à deux
dimensions, en vue de sonder des processus vibrationnels et électroniques [1]. Comme en RMN,
une approche de spectroscopie par transformée de Fourier permet de résoudre le spectre
d’excitation d’un système, tout en mesurant simultanément son spectre de transmission, afin
d’établir des corrélations révélant d’éventuels couplages. La mise en oeuvre expérimentale peut
ainsi être réalisée en insérant un simple interféromètre sur le bras de pompe d’une expérience
pompe‐sonde [2]. Après une description des concepts à la base de la spectroscopie
multidimensionnelle et de la mise en oeuvre expérimentale dans le cas du domaine infrarouge,
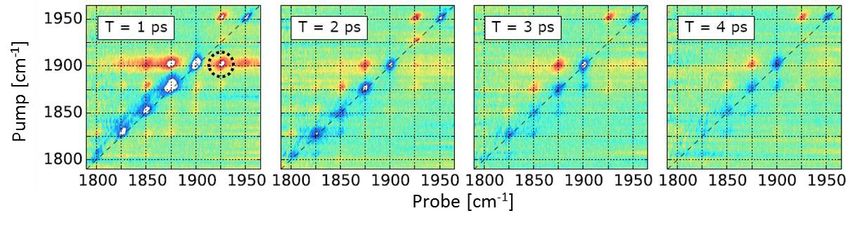
nous présenterons une application à l’étude de la carboxy‐hémoglobine [3, 5].
Fig. 1. Spectre 2DIR obtenu dans HbCO (à gauche) comparé à une simulation numérique (à droite).
Extrait de [3].
D’une part, nos résultats (Fig. 1) montrent un très bon accord entre la mesure expérimentale à l’équilibre
et une méthode numérique hybride quantique/classique permettant de rendre compte de l’effet des
fluctuations structurelles de la protéine sur la réponse infrarouge du ligand considéré [3]. D’autre part,
en combinaison avec une excitation préalable du système par ascension vibrationnelle [4], la
spectroscopie 2DIR hors d’équilibre constitue une nouvelle méthode d’étude de la relaxation
vibrationnelle dans des régions largement inexplorées de la surface de potentiel (Fig. 2).
Fig. 2. Série de spectres 2DIR mesurés dans une échelle vibrationnelle hors d’équilibre peuplée par
ascension vibrationnelle à l’aide d’une impulsion infrarouge à dérive de fréquence, pour différentes
valeurs du retard T entre la dernière impulsion de pompe et l’impulsion sonde. Les différents pics sont
répartis sur une grille en raison de l’anharmonicité du potentiel, ce qui permet d’identifier les transitions
vibrationnelles impliquées. Le changement de signe des pics observés à droite de la diagonale illustre la
relaxation vibrationnelle des différents niveaux. Extrait de [5]
25[1] J. P. Ogilvie, K. J. Kubarych, Adv. At. Mol. Opt. Phys. 57, 249 (2009).
[2] L. P. DeFlores, R. A. Nicodemus, A. Tokmakoff, Opt. Lett. 32, 2966‐2968 (2007).
[3] C. Falvo, L. Daniault, T. Vieille, V. Kemlin, J.‐C. Lambry, C. Meier, M.H. Vos, A. Bonvalet, M. Joffre, J. Phys.
Chem. Lett. 6, 2216‐2222 (2015).
[4] C. Ventalon, J.M. Fraser, M.H. Vos, A. Alexandrou, J.‐L. Martin, M. Joffre, Proc. Natl. Acad. Sci. USA 101, 13216
(2004).
[5] V. Kemlin, A. Bonvalet, L. Daniault, M. Joffre, J. Phys. Chem. Lett. 7, 3377‐3382 (2016)
26The photochemistry of inverse dithienylethene switches understood
S. Aloïse
LASIR Lille
27Alignement moléculaire par impulsions laser ultra‐brèves : production, mesure et
applications récentes
E. Hertz
ICB Dijon
L’interaction entre un échantillon moléculaire et une impulsion laser ultra‐brève intense
conduit, après le passage de l’impulsion, à des réalignements périodiques et transitoires des
molécules. Parmi les différentes stratégies de contrôle développées ces dernières années,
l’alignement moléculaire dans ce régime post‐impulsionnel ou field–free a fait l’objet d’un intérêt
particulièrement important. Sa première mise en évidence expérimentale en 2001 [1] a été
accompagnée de nombreux travaux ayant reporté son utilisation dans des domaines
d’applications très variés : diagnostiques optiques, réactions photo‐chimiques, optique non
linéaire, filamentation laser… Après une brève introduction du phénomène, l’exposé se
focalisera sur deux champs d’investigation explorés actuellement dans l’équipe: la production
de rotation moléculaire unidirectionnelle [2] et le contrôle de la polarisation de champs
harmoniques [3].
[1] F. Rosca‐Pruna, M. J. J. Vrakking PRL 87, 153902‐1 (2001)
[2] G. Karras, M. Ndong, E. Hertz, D. Sugny, F. Billard, B. Lavorel, O. Faucher PRL 114, 103001(2015)
[3] J. Houzet, E. Hertz, F. Billard, B. Lavorel, and O. Faucher, Phys. Rev. A 88, 023859 (2013)
28Spectroscopie de cohérences vibrationnelles et photoréactivité en phase liquide
Moussa Gueye, Stefan Haacke, Jérémie Léonard.
Institut de Physique et Chimie des Matériaux de Strasbourg & Labex NIE, Université de
Strasbourg, CNRS UMR 7504, Strasbourg, France
Jeremie.Leonard@ipcms.unistra.fr
La photoisomérisation d’une double liaison C=C est une photoréaction qui convertit l’énergie
lumineuse en énergie mécanique à l’échelle moléculaire. Dans la rhodopsine (Rho),
photodétecteur de la vision chez les vertébrés, la photoisomérisation du rétinal, qui active la
fonction biologique, est très rapide et son rendement quantique est élevé (67%). Il est
remarquable que cette photoréaction est également vibrationnellement cohérente. Cela signifie
que le mouvement de rotation autour de la double liaison est balistique, décrit par un paquet
d’onde vibrationnel qui conduit le système moléculaire à travers une « intersection conique »
entre les surfaces d’énergie potentielle électronique des états excité et fondamental
(photoproduit). La conversion photomécanique se fait sur une échelle de temps plus rapide que
la dissipation d’énergie vers le bain thermique et le rendement est contrôlé par la phase de
certains modes vibrationnels au passage de l’intersection conique.[1]
Nous étudions le mécanisme de photoisomérisation, en solution, de composés synthétiques
dérivés du « N‐alkylated indanylidene–pyrrolinium » (NAIP), dont la structure électronique est
inspirée du rétinal dans la Rho.[2] Certaines de ces molécules reproduisent une photoréaction
très rapide et vibrationnellement cohérente, mais n’ont un rendement que de 20 à 35%. [3, 4]
Tout comme les protéines de rétinal elles sont des systèmes moléculaires modèles pour étudier
la photophysique des intersections coniques au voisinage desquelles l’approximation de Born‐
Oppenheimer s’écroule et la transition électronique entre l’état excité et le photoproduit est
contrôlée par la dynamique vibrationnelle.
Nous avons récemment construit un nouveau montage d’absorption transitoire [5] qui utilise
une impulsion sub‐8fs, centrée à 400 nm, comme pompe permettant d’exciter impulsivement
des paquets d’onde vibrationnels dans tous les états électroniques optiquement couplés. Un
supercontinuum de lumière blanche (300‐900nm) est utilisé pour sonder les signatures de ces
paquets d’ondes au cours de la photoréaction. Il s’agit donc d’une spectroscopie de cohérences
vibrationnelles qui est en réalité une spectroscopie Raman réalisée dans le domaine temporel
plutôt que spectral.
Grâce à cette expérience nous avons pu apporter la preuve expérimentale définitive que la
photoréaction de certains des composés NAIP est vibrationnellement cohérente: un paquet
d’onde vibrationnel, créé par le mouvement réactionnel sur la surface de l’état excité, conduit le
système à travers l’intersection conique, et continue d’osciller dans l’état fondamental du
photoproduit avant complète dissipation (1. Schapiro, I., M.N. Ryazantsev, L.M. Frutos, N. Ferre, R. Lindh, et al., ʺThe Ultrafast Photoisomerizations of
Rhodopsin and Bathorhodopsin Are Modulated by Bond Length Alternation and HOOP Driven Electronic
Effectsʺ. J. Am. Chem. Soc. 133, 3354‐3364 (2011).
2. Sinicropi, A., E. Martin, M. Ryazantsev, J. Helbing, J. Briand, et al., ʺAn artificial molecular switch that mimics
the visual pigment and completes its photocycle in picosecondsʺ. Proc. Nat. Acad. Sci. USA. 105, 17642‐17647
(2008).
3. Briand, J., O. Braem, J. Rehault, J. Leonard, A. Cannizzo, et al., ʺCoherent ultrafast torsional motion and
isomerization of a biomimetic dipolar photoswitchʺ. Physical Chemistry Chemical Physics. 12, 3178‐3187 (2010).
4. Léonard, J., I. Schapiro, J. Briand, S. Fusi, R.R. Paccani, et al., ʺMechanistic Origin of the Vibrational Coherence
Accompanying the Photoreaction of Biomimetic Molecular Switchesʺ. Chem. A Eur. J. 18, 15296‐15304 (2012).
5. Gueye, M., J. Nillon, O. Crégut and J. Léonard, ʺBroadband UV‐Vis vibrational coherence spectrometer based on
a hollow fiber compressorʺ. Rev. Sci. Instrum. 86, (2016), in press, DOI: 10.1063/1.4962699
30Time‐domain investigations of the vibrational and cooling dynamics of metal and hybrid
nano‐objects
Aurélien Crut, Tatjana Stoll, Denis Mongin, Vincent Juvé, Paolo Maioli, Fabrice Vallée and
Natalia Del Fatti
FemtoNanoOptics Group, institut Lumière Matière, CNRS‐Université Lyon 1, Université de Lyon,
Villeurbanne, France
Nano‐objects exhibit discrete vibrational modes, which can be monitored in the time
domain using ultrafast optical pump‐probe spectroscopy. Their study enables fundamental
investigations of acoustics at the nanoscale, characterization of the mechanical response of
nano‐objects and design of artificial nanoresonators with frequencies reaching the THz domain.
The most striking result of experiments performed on assemblies of small metal nanoparticles
or clusters has been the quantitative agreement of the experimentally measured vibrational
periods with those computed with a macroscopic elastic model, using bulk elastic constants, a
result subsequently confirmed by atomistic simulations. The effect of an inhomogeneous nano‐
object environment has also been addressed both experimentally and theoretically by
considering spherical or elongated nano‐objects encapsulated in metal or dielectric shells.
Additionally, understanding of vibrational damping is currently emerging thanks to the
realization of time‐resolved spectroscopy experiments on single nanoparticles, which avoids the
spurious inhomogeneous effects affecting ensemble measurements.
Time‐resolved signals acquired on colloidal or glass‐embedded metal nanoparticles also
present a signature of their thermal dynamics, i.e. initial heating by the ultrafast pump pulse
and subsequent cooling by energy transfer to the environment. The cooling dynamics of such
nanoparticles fundamentally differ from those of macroscopic objects due to the increased role
played by Kapitza interface resistances. These thermal resistances can be deduced from the
analysis of time‐resolved signals, at the condition of carefully quantifying the environment
contribution to these signals and using a complete thermal model accounting for both heat
transfer at the particle‐environment interface and heat diffusion in the surroundings.
31From molecular switching to material transformation: revisiting the spin crossover
Maciej Lorenc
Institut de Physique de Rennes (IPR), Université de Rennes 1, France ,
maciej.lorenc@univ‐rennes1.fr
The general excitement born out of the ultrafast clock caused rush of new ideas, new
materials, and new instruments. One feat in particular has been draining a lot of effort, namely
the control of materials with an ultrashort laser pulse. There is ample evidence now that
materials can be directed between different macroscopic states by using appropriate electronic,
or structural, excitations. The switching with a laser pulse of such materials can severely change
their macroscopic properties (electric conductivity, magnetism, colour, etc.), whereby emerging
cooperativity and coherence of different degrees of freedom underpin the resulting phase
transitions of various sorts. However, the pertinent time scales for photo‐switching processes in
materials have been rather difficult to scrutinise. The pioneering investigations dealt mainly
with the electron/phonon dynamics immediately following the femtosecond excitation, or the
kinetics of recovery to the thermally stable states. Time‐resolved X‐ray diffraction and ultrafast
VIS‐IR spectroscopy reveal that the degrees of freedom triggered by a femtosecond laser pulse
in a spin‐crossover (SCO) material follow a sequence in the out‐of‐equilibrium dynamics. Those
steps dissected in time, provided a mechanistic picture of a material transformation driven
under different regimes (coherent or stochastic).
[1] Lorenc M. et al., Successive Dynamical Steps of Photoinduced Switching of a Molecular Fe(III) Spin‐Crossover
Material by Time‐Resolved X‐Ray Diffraction, Phys. Rev. Lett. 103, 028301 (2009)
[2] Cailleau H. et al., Impacting Materials by Light and Seeing their Structural Dynamics, Eur. Phys. J. Special
Topics 222, 1077 (2013)
[3] Cammarata M. et al., Sequential Activation of Molecular Breathing and Bending during Spin‐Crossover
Photoswitching Revealed by Femtosecond Optical and X‐Ray Absorption Spectroscopy, Phys. Rev. Lett. 113,
227402 (2014)
[4] Bertoni R. et al., Elastically Driven Cooperative Response of a Molecular Material Impacted by a Laser Pulse,
Nature Mater. 15, 606 (2016)
32Dynamical Disorder in the DNA Hydration Shell
C. Hynes
PASTEUR‐ENS Paris
33Out‐of‐equilibrium electron dynamics in topological matter
E. Papalazarou1, L. Khalil12, M. Caputo1, N. Nilforoushan1, L. Perfetti3, M. Marsi1
1Laboratoire de Physique des Solides, CNRS, Univ. Paris‐Sud, Université Paris‐Saclay, 91405 Orsay
Cedex, France
2Synchrotron SOLEIL, Saint Aubin BP 48, Gif‐sur‐Yvette F‐91192, France
3Laboratoire des Solides Irradiés, Ecole Polytechnique, CNRS, CEA, Université Paris‐Saclay, 91128
Palaiseau cedex, France
In the recent years, the topological phases of matter have been in focus of intense theoretical
and experimental studies. The generic features of these phases are: they possess topologically
protected surface states and their order can be described by topological invariants that are
insensitive to smooth changes in the system parameters.
Among these phases three‐dimensional topological insulators exhibit a great attention because
they possess a band gap in the bulk and conducting surface states. Carriers occupying these
surface states are highly protected by time‐reversal symmetry leading to a substantial
suppression of back‐scattering and to a robust helical spin texture.
Using femtosecond time‐ and angle‐resolved photoelectron spectroscopy we reveal the out‐of‐
equilibrium electron dynamics of topologically protected surface states. We show that the
interplay of surface and bulk transient carrier dynamics in a photo‐excited topological insulator
compound may alter the balance between electrons and holes in Dirac surface states. This can
result to an unusually long‐lived population of hot massless Dirac fermions following
femtosecond optical photo‐excitation.
34Champ fort au bout d’une pointe: nouveaux outils pour la photoémission et le contrôle
cohérent.
B. Chalopin
LCAR Toulouse
L’interaction d’impulsions laser ultra‐courtes avec des nanopointes métalliques permet
d’étudier plusieurs régimes de photoémission d’électrons qui peuvent être mis en évidence
grâce à leur signature spectrale. En utilisant des nanopointes de différents matériaux, nous
avons observé le régime d’émission multiphotonique au‐dessus du seuil ainsi que l’émission de
champ optique avec recollision des électrons. Cette étude a notamment permis de caractériser
l’amplification du champ optique dans une nanopointe d’argent. À cause du caractère
plasmonique du matériau, ce facteur d’amplification (défini comme le rapport entre l’intensité
optique locale et l’intensité des impulsions incidentes) dépend de la longueur d’onde. La
polarisation permet également de contrôler le champ optique à l’apex de la pointe et permet
donc de contrôler le profil spatial d’émission. À la différence de la phase gazeuse, les
nanopointes sont sensibles à des effets thermiques en fonction du taux de répétition des
impulsions lasers. Pour étudier cela, nous avons développé une source laser NOPA accordable
en longueur d’onde et en taux de répétition.
35Vous pouvez aussi lire

















































