PROGRAMME 2017-2018 - Cours Pasteur
←
→
Transcription du contenu de la page
Si votre navigateur ne rend pas la page correctement, lisez s'il vous plaît le contenu de la page ci-dessous
Cours Pasteur
ANALYSE DES GENOMES
PROGRAMME
2017-2018
1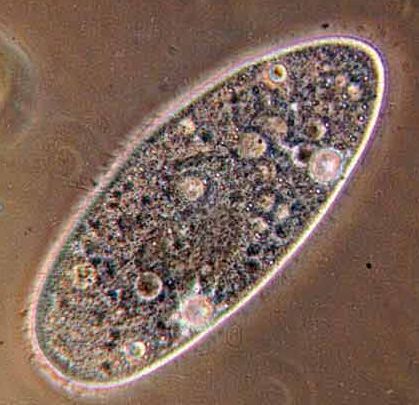
Cours Pasteur
ANALYSE DES GENOMES
2017-2018
******
Codirecteurs du Cours :
Stéphane LE CROM & Didier MAZEL
Chef de Travaux :
Lionel FRANGEUL
******
LE COURS SE DEROULE DU 6 NOVEMBRE AU 21 DECEMBRE 2017
AU CENTRE D’ENSEIGNEMENT DE L’INSTITUT PASTEUR
(PAVILLON LOUIS MARTIN, BATIMENT 09)
28, RUE DU DOCTEUR ROUX, 75015 PARIS
CONFERENCES ET COURS :
- DU 6 NOVEMBRE AU 21 DECEMBRE 2017 :
SALLE DE COURS 3 (BATIMENT SOCIAL 06)
TRAVAUX PRATIQUES : SALLE DE TP 2EME ETAGE DU CENTRE D’ENSEIGNEMENT
(PLM, BATIMENT 09)
2
PRESENTATION DU COURS
Préambule au cours d’Analyse des Génomes 2017-2018
La Génétique est la Science qui étudie l’hérédité. Or, quiconque s’interroge sur les différences
entre un objet physique, par exemple un nuage, et un organisme vivant, par exemple une souris,
arrivera tôt ou tard à la conclusion inévitable qu’il n’y en a qu’une: l’hérédité car, comme les nuages,
les organismes vivants suivent les lois de la physico-chimie (voir Schrödinger, 1944). Ils sont
constitués des mêmes atomes. Mais, alors qu’un nuage se forme à une date et en un lieu donnés
comme la conséquence d’un ensemble de valeurs précises d’humidité, de pression et de température
sans souvenir de la présence éventuelle d’un autre nuage, similaire ou non, à une date antérieure, une
souris naît à partir de deux autres souris préexistantes qui, elles-mêmes, avaient des parents, etc …
Pour sa formation à partir des atomes et des molécules qui la constitueront, une souris hérite, dès
l’oeuf, du fruit de l’évolution de tous ses ancêtres, proches et lointains, tandis que le nuage part de
zéro. Les êtres vivants ont donc, en plus de la physique, une histoire portée de génération en
génération par le matériel héréditaire. Connaître ce matériel héréditaire et son fonctionnement c’est
donc lire l’histoire des êtres vivants, comprendre leur complexité et, finalement, appréhender ce qui les
distingue du monde inanimé. C'était toujours l’objet même de la Génétique depuis son origine même si
les méthodes d’analyse n’ont longtemps permis de lever que quelques pans du voile. Avec l’analyse
des génomes, notre connaissance du matériel héréditaire devient exhaustive et, s’éloignant
progressivement des systèmes modèles qui furent si précieux à la Génétique, la Génomique explore
maintenant le monde vivant dans son intégralité et, progrès techniques aidant, à travers tout le
spectre d'échelles qui relie les molécules élémentaires aux populations naturelles. Des horizons
insoupçonnés se découvrent. Les notions classiques font place à des visions nouvelles qui nous
permettent même d’imaginer des mondes que la Biologie synthétique essai de construire. Pour bien
appréhender ces idées, un bref retour en arrière s’impose. On mesurera d’autant mieux
l’impressionnant chemin parcouru que l’année 2015 marquera le 150ème anniversaire de la présentation
des travaux de Mendel (Mendel, 1866),
La Génétique, science des génomes
Les bases de la génétique moléculaire
Au cours du siècle dernier, nos connaissances sur le matériel héréditaire ont progressé d’une
manière considérable. Depuis les chromosomes eucaryotes, corpuscules observables au microscope
au cours des divisions cellulaires dont le comportement trahissait leur rôle dans l’hérédité pour ceux
qui connaissaient les lois de Mendel, on est passé à l’ADN grâce aux bactéries (Avery et al., 1944,
Watson et Crick, 1953). Puis, on a décrit la structure fine du gène grâce aux bactériophages (Benzer,
1961) et déchiffré le code génétique grâce essentiellement à la Biochimie (Crick et al., 1961,
Nirenberg et al., 1961, Nishimura et al.,1965). Avec les opérons bactériens, on découvrait des
principes de régulation de l'expression des gènes qui semblaient universels (Jacob et Monod, 1961).
On savait, grâce aux champignons, qu’à chaque gène correspondait une protéine (Beadle et Tatum,
1941). Et le dogme central de la Biologie moléculaire (datant de 1953, voir figure 1) nous indiquait
comment les ARN, jouant le rôle d'intermédiaires, étaient impliqués dans l'expression des gènes pour
former ces protéines. Nul ne doutait alors que ces principes étaient universels et certains, pensant que
l'on avait compris l'essentiel, se détournèrent à ce moment de la biologie moléculaire des gènes pour
3
s'intéresser au développement des organismes, au fonctionnement du système nerveux ou à d'autres
problématiques jugées plus complexes.
Les ARN: pas uniquement de simples messagers
Pourtant, la Génétique moléculaire devait révéler encore bien d'autres surprises sans lesquelles
l’analyse des génomes aujourd’hui serait incompréhensible. D'abord, on découvrit que les ARN
peuvent être retrotranscrits sous forme d'ADN pouvant être intégré au matériel génétique et donc
transmis à la descendance (Temin et Mizutani, 1970, Baltimore, 1970). Dès lors, les ARN n'étaient
plus seulement des intermédiaires de l'expression des gènes, ils pouvaient donner naissance au
matériel héréditaire. Ensuite, dès que l'on a pu étudier directement la structure moléculaire des
gènes, grâce aux techniques de l’ADN recombinant et du Génie génétique (développées à partir de
1973), celle-ci est immédiatement apparue beaucoup plus complexe qu'on ne l'imaginait. Et même
surprenante. On découvrit les introns, séquences internes des ARN transcrites de l'ADN mais
éliminées des molécules d'ARN finales par épissage des séquences qui les entourent, les exons
(Berget et al. 1977, Chow et al., 1977, Glover et Hogness, 1977, Jeffreys et Flavell, 1977, Gilbert,
1978). On parlait de gènes mosaïques que l’on commençait à séquencer en essayant toujours
d’interpréter les résultats selon les principes du dogme central de la biologie moléculaire.
En réalité, on était en train de mettre en lumière le rôle central des ARN, les gènes n’en étant
que le reflet. On sait maintenant qu'il existe plusieurs catégories d'introns et les différents mécanismes
de l'épissage des ARN ont été décrits. On découvrit que, dans la plupart des cas, ce sont les ARN eux-
mêmes qui catalysent ces réactions d’épissage (voir plus loin) même si, pour ce faire, ils sont parfois
associés à des protéines. Sans entrer dans les détails pourtant très significatifs, l'idée importante ici est
qu'entre le gène et son produit s'intercalent une série de réactions qui modifient, souvent
considérablement, les séquences des populations de molécules d'ARN présentes dans la cellule. Or,
pour des raisons techniques, c’est le séquençage de l’ADN qui s’est développé et non celui de l’ARN
dont les molécules chimiquement très réactives permettent mal le séquençage direct (sans faire une
copie ADN). Actuellement, le séquençage massif d’ARN (voir plus loin) passe par un intermédiaire
ADNc.
Les débuts du séquençage de l’ADN
Les premières méthodes qui permirent de déterminer rapidement l'ordre de succession des
nucléotides le long des molécules d'ADN (séquencer l'ADN) datent de 1977 (Sanger et al., 1977,
Maxam et Gilbert, 1977). C’est une date critique. Avant, on savait conceptuellement ce que devait être
un gène et ses mutations, mais sans espoir d’en connaître réellement le contenu informatif précis.
Après, on allait pouvoir déchiffrer ce contenu, vieux rêve de tous les généticiens. Ces méthodes sont
aujourd'hui reléguées aux musées (voir plus loin), mais il s'agissait alors d'un progrès considérable qui
faisait suite à des années de recherches au cours desquelles avaient été explorées différentes pistes
permettant de déterminer des séquences courtes d’ADN comme, par exemple, les opérateurs
bactériens. Ce n’est donc qu’à partir de 1977 que l’on a commencé à connaître l'information génétique
contenue dans les gènes. Une accélération considérable des découvertes de la génétique moléculaire
s'ensuivit. Les mutations n'étaient plus uniquement des signatures conceptuelles associées à des
phénotypes particuliers dans des conditions définies du laboratoire. On en découvrait maintenant la
nature chimique et, en conséquence, on allait pouvoir les créer chimiquement de façon déterminée.
Toute l'histoire de la mutagénèse dirigée débutait, suivie plus tard de celle de la synthèse chimique
des gènes et maintenant de celle des génomes entiers (voir plus loin).
4Comme le dogme central de la biologie moléculaire et le code génétique permettent de prédire
les séquences des protéines à partir de celles des gènes (aux modifications près introduites au niveau
des ARN), au début des années 1980s on séquençait les gènes pour avoir la séquence des protéines.
Mais le séquençage d’ADN restait laborieux et le souci était d’éviter la duplication des efforts.
Naissaient alors les premières bases de données permettant de mettre à la disposition de la
communauté scientifique les séquences d'ADN et celles, déduites, de protéines. Peu à peu, comme
ces répertoires s'enrichissaient, les comparaisons de séquences devenaient possibles. Graduellement,
elles allaient prendre le pas sur les expériences. En même temps, on s'intéressait aux séquences
régulatrices de l'expression des gènes que l'on pouvait maintenant manipuler dans des systèmes
artificiels d'expression génétique. On s'intéressait évidemment aussi aux premiers gènes morbides
identifiés chez l'homme. On espérait en tirer rapidement des traitements (et des retombées
financières !). On s'intéressait aux génomes des organelles, des plasmides et des virus dont les tailles
limitées permettaient d'obtenir les séquences complètes en seulement …. quelques années de travail
! C’était l’époque du Génie génétique triomphant. Certains, pensant alors que l’on avait tout compris,
ne rêvaient que d’applications. Elles furent décevantes pour la plupart car très prématurées.
L’ingénierie génomique
C'est pourtant à cette époque que furent découverts les premiers outils d'ingénierie des
génomes. Des endonucléases dont la spécificité de séquence permettait d'envisager cibler un site
unique dans un génome entier. La première catégorie d'enzymes de cette nature, appelée maintenant
homing endonucleases, avait été découverte à partir d' un intron mobile d'un gène mitochondrial de
levure présentant des anomalies de transmission héréditaire lors des croisements (Jacquier et Dujon.,
1985, Colleaux et al., 1986, Colleaux et al. 1988). Il s’agissait de l’aboutissement totalement imprévu
de plus de quinze ans de recherches sur un phénomène surprenant dont le seul intérêt était son
existence même (Dujon, 2005a), tout sauf le chemin direct avec rapports d’étapes souhaité par les
tenants actuels de la recherche sur projets prédéfinis répondant à un défi sociétal ! A cette époque,
grâce au CNRS et à la liberté qui régnait dans les universités françaises, on pouvait découvrir ce que
l’on ne cherchait pas, simplement parce que c’était possible de le faire, un peu comme on vainc le
sommet d’une montagne. De très nombreuses homing endonucleases ont ensuite été découvertes
issues d'une variété d'organismes ou ont été synthétisées artificiellement pour des applications
précises. Plus tard (1996) est apparue une deuxième catégorie d'endonucléases site-spécifiques dites
« à doigt de zinc ». Le principe en était très différent puisque, au lieu d’exploiter ce que fournissait la
nature, il s’agissait d’une ingénierie moléculaire, joignant une endonucléase bactérienne classique à
une combinaison de motifs protéiques élémentaires capable de conférer à l’ensemble une
reconnaissance spécifique d’un fragment de séquence suffisamment long pour assurer son unicité
dans un génome. Sur le même principe général, sont nées plus récemment (2010) une troisième
catégorie d'endonucléases site-spécifiques appelées TALE nucleases ou TALEN. Enfin, très
récemment (2012) une quatrième catégorie d'endonucléases site-spécifiques a pu être développée à
partir d’observations faites chez des bactéries chez lesquelles une « immunité » antiphagique est
obtenue par activation d’une endonucléase protéique non-spécifique par un petit ARN qui, lui, permet
la reconnaissance parfaite d’un fragment de séquence suffisamment long pour assurer son unicité
dans un génome. La facilité avec laquelle on peut synthétiser artificiellement des petits ARN,
comparée à la synthèse de protéines recombinantes, a assuré un enthousiasme immédiat pour ce
dernier système appelé CRISPR. Avec ces outils, on entre dans une nouvelle ère de Génie
génomique qui ouvre les plus grands espoirs (voir figure 2).
Les multiples fonctions des ARN
5Pendant ce temps, les ARN continuèrent de nous surprendre. D'abord, on découvrit qu'ils
subissent des éditions, c'est-à-dire que leur séquence est modifiée de façon précise et déterminée,
changeant ainsi l'information génétique qu'ils étaient censés véhiculer. On connaît maintenant
beaucoup de mécanismes différents d'édition. Dans certains cas, l'édition peut être tellement massive
qu'elle crée des messagers traduits en protéines là où il n'y a pas de gène reconnaissable
correspondant. C’est le cas des mitochondries dans le grand groupe eucaryote des Excavates (voir
figure 3). Mais surtout on découvrit que les ARN sont capables de catalyser des réactions chimiques
(Cech et al., 1981, Altman, 1981). D’abord celles concernant leur propre structure
(transesterifications permettant l'épissage des introns, hydrolyse des liaisons phosphodiester
permettant la maturation des ARN précurseurs). Mais aussi toute une variété d'autres réactions
biochimiques. Aujourd'hui on sait que les ARN sont impliqués, comme catalyseurs ou comme co-
facteurs, dans une variété de réactions essentielles à la vie cellulaire telles que la synthèse protéique
au niveau du ribosome, l'élongation des télomères (Greider and Blackburn, 1989), le transport des
protéines, les processus de maturation ou de modifications chimiques d'autres ARN et, bien sûr, le
contrôle de l’expression d’autres gènes ainsi que des éléments mobiles, des séquencs virales ou des
séquences répétées dans les génomes. On découvrit des machineries complexes chez les eucaryotes,
impliquant des petits ARN, pour ces dernièrs types d’activités (Fire et al., 1998). Le nombre des petits
ARN et la variété de leurs propriétés ont augmenté très vite grâce, en particulier, aux nouvelles
méthodes de séquençage.
Le séquençage des génomes et le développement de la Génomique
Les motifs
Au milieu des années 1980s, les applications potentielles du génie génétique et d’autres
considérations plus stratégiques, voire politiques, allaient motiver le séquençage des génomes entiers,
à commencer par celui de l'homme. Plusieurs années s'ensuivirent au cours desquelles hésitations,
conflits et rebondissements ne furent pas rares. Contrairement aux idées simples, les progrès les plus
décisifs ne vinrent pas toujours de là où on les attendait. Comme dans toute recherche véritable
d'ailleurs. Des bactéries (comme Haemophilus influenzae), la levure de boulangerie Saccharomyces
cerevisiae et le nématode Caenorhabditis elegans devaient jouer, chacun à leur manière, des rôles
essentiels dans le programme "génome humain" alors qu'ils étaient des initiatives indépendantes (lire,
par exemple, Vassarotti et al.,1995, Goujon, 2001, Brown, 2003). Ironiquement, alors que certains ne
voyaient dans ces génomes que des tremplins technologiques pour le génome humain, c’est sur le
plan conceptuel que les choses commençaient à bouger.
Les surprises
Les premiers génomes séquencés (Fleischmann et al. 1995, Goffeau et al. 1996) nous
rappelèrent rapidement à quel point des connaissances fondamentales nous manquaient. Avec le
génome de la levure, trois surprises majeures attendaient les généticiens. D’abord, il y avait dans le
génome beaucoup plus de gènes pour chaque fonction que ce que la génétique laissait prévoir. En
d’autres termes, les cribles génétiques classiques mêmes les plus systématiquement appliqués
n’arrivaient jamais à l’exhaustivité. Ensuite, beaucoup de gènes avaient des séquences entièrement
nouvelles, sans similarité dans les bases de données existantes. Une explication triviale était que ces
bases de données étaient très incomplètes, ce qui n’était pas faux. Mais même aujourd’hui chaque
nouveau génome séquencé fait apparaître une fraction non nulle de tels gènes qu’on désigne donc
comme « orphelins ». Une autre explication commune à l’époque était que ces gènes orphelins
n’étaient pas des vrais gènes. Ce qui n’est pas nécessairement faux non plus pour certain d’entre eux.
6Mais leur nombre élevé exclu la généralisation de cette hypothèse. Une réalité plus intéressante,
comprise seulement maintenant, est que certains des gènes orphelins sont en réalité des gènes créés
de novo dans les différentes lignées évolutives. Enfin, la troisième surprise était que nombre de gènes
étaient dupliqués. Ceci était incompréhensible dans la vision classique de mutations aléatoires
soumises à la sélection naturelle. On sait maintenant que cette redondance est vraie pour tous les
génomes, même si le cas de la levure était particulier. En d'autres termes, la nature ne connaît pas les
génomes minimums dont rêvent les ingénieurs. La raison est à rechercher dans la perpétuelle
dynamique évolutive des génomes (voir plus loin).
Les chiffres
Actuellement, de nombreux génomes bactériens ont été séquencés entièrement ou
partiellement (plus de 219 000 projets sont mentionnés sur le site GOLD
(http://www.genomesonline.org/). Il en va de même d'environ deux mille génomes d'Archaea (un déficit
important comparé aux bactéries) et d'un nombre rapidement croissant d’eucaryotes (environ 14 500
sont terminés ou en cours). Historiquement, ce fut la levure Saccharomyces cerevisiae avec son
génome d'environ 13 millions de nucléotides (Mb) le premier eucaryote séquencé (Goffeau et al. 1996,
1997). Puis, alors que le nombre de génomes bactériens augmentait, on a vu apparaître
successivement les séquences de génomes eucaryotes plus grands tels que ceux de Caenorhabditis
elegans, (97 Mb, Sulston, Waterston et Consortium, 1978), un nématode servant de modèle
expérimental, et d'Arabidopsis thaliana (115 Mb, Arabidopsis Genome Initiative, 2000), une crucifère
modèle. Ces débuts étaient très laborieux. Ils nécessitaient plusieurs années de travail de consortiums
de laboratoires qui établissaient d'abord une cartographie détaillée des génomes avant un séquençage
ordonné des segments par la méthode de Sanger. Chacun de ces projets marquait une étape
importante de la génomique naissante.
Le génome humain
Au tournant de l'an 2000, un premier assemblage du génome de Drosophila melanogaster
(160 Mb) était publié, démontrant la faisabilité d'un séquençage aléatoire total, dit shotgun (Adams et
al., 2000). Il s'agissait d'une étape importante dans la course au génome humain. Celui-ci (environ
3,100 Mb) a été déclaré terminé dans une première version en 2001 (Collins et al., 2001, Venter et al.,
2001). C'était un travail considérable qui avait impliqué pour l'International Human genome
Consortium, le séquençage chromosome par chromosome, par l'intermédiaire de clones BAC ancrés
sur une cartographie génétique, et qui s'est terminé par une compétition contre un groupe privé
travaillant par séquençage total aléatoire. Compétition biaisée car, alors que les séquences
chromosome par chromosome du Consortium international étaient rendues immédiatement publiques,
celles du groupe privé restaient confidentielles. Une version plus complète et révisée du génome
humain fut publiée par l'International Human genome Sequencing Consortium (2004). Il s'agissait
toujours d'un "génome théorique", c'est-à-dire d'un équivalent haploïde de plusieurs individus. Depuis,
les génomes de plusieurs personnes vivantes ont été séquencés et certains scientifiques connus ont
souhaité voir leurs génomes publiés les premiers. Après plusieurs autres génomes de représentants
de différentes populations ayant permis les premières comparaisons, un vaste projet d'étude du
polymorphisme a été lancé impliquant le séquençage de plus d'un millier d'individus appartenant à 14
populations (The 1000 Genomes Project Consortium, Abecassis et al., 2012). Avec les génomes
individuels, on découvre qu'au-delà des SNPs et indels, le polymorphisme génétique entre les
individus implique de grandes variations structurales dont l'importance était sous-estimée, telles que de
larges délétions, duplications ou inversions (Korbel et al., 2007) et des réarrangements balancés
(Chen et al., 2008). Les variations du nombre de copies de segments de chromosomes (CNVs) sont
7maintenant reconnues comme une source majeure de polymorphisme des génomes et il est
maintenant devenu clair que, chez les eucaryotes au moins, les altérations chromosomiques
dépassent en fréquence les mutations ponctuelles. L'analyse des données de polymorphisme est en
train de nous apporter de nombreuses informations sur les variations entre individus (Abecassis et al.,
2012), l'origine des indels (Montgomery et al., 2013), les évènements de rétroduplications (Abyzov et
al., 2013) ou encore les variations fonctionnelle d'expression des gènes (Lappalainen et al., 2013) pour
ne citer que quelques exemples. Des espoirs considérables apparaissent dans le domaine des
cancers (Khurana et al., 2013) en particulier grâce à la possibilité d'identifier des allèles à faible
pénétrance (Whiffin et al., 2013).
Les grands génomes
Après la première version du génome humain apparurent les génomes d'autres vertébrés qui
devaient jouer un rôle fondamental dans l'interprétation du génome humain. Il s'agit du Fugu (365 Mb,
Aparicio et al., 2002), un poisson téléostéen, et de son cousin Tetraodon negroviridis (Jaillon et al.,
2004). C'est avec ce dernier que, par comparaison détaillée, l'on réussit à déduire que le génome
humain devait compter seulement 23,000 gènes environ. Vinrent aussi les génomes du riz (420-466
Mb, Goff et al. 2002, Yu et al., 2002, Yu et al., 2005), d’Anopheles gambiae (278 Mb, Holt et al., 2002),
un moustique vecteur de la malaria, d'autres nématodes (Stein et al., 2003, Mitreva et al., 2005), de la
souris (Waterston et al., 2002, Mouse genome consortium, 2002), du rat (Gibbs et al., 2004), du poulet
(Hillier et al., 2004), du chimpanzé (Mikkelsen et al., 2005) et d'autres grands primates. Ensuite,
apparurent les génomes du peuplier, du chien, de la vigne, du cheval, du bananier, de
l'ornithorhynque, du concombre, de la papaye, du ver à soie pour ne citer que quelques exemples
datant encore de la période Sanger (voir plus loin). Il est devenu impossible de suivre cette
accélération. Malgré cette abondance, chaque nouveau génome continue de nous révéler des
surprises. Tous ces génomes ne sont pas nécessairement séquencés de manière complète. À cause
de leur taille même, ou des difficultés inhérentes à leur complexité, on réalise le séquençage à un
certain niveau de couverture moyenne 1, variable selon les besoins. Il reste des trous ou des zones de
basse qualité dans les séquences déposées dans les bases de données. Il faut s'en souvenir même si
les progrès de la Génomique comparée permettent de s'en accommoder. Et surtout les méthodes de
séquençage ayant considérablement évolué (voir plus loin), les problèmes se posent aujourd’hui de
manière totalement différente pour les nouveaux génomes étudiés.
La génomique évolutive
En parallèle des grands génomes cités, le séquençage total ou partiel de beaucoup d'autres
génomes eucaryotes de taille plus modeste était devenue chose courante au début des années 2000
en utilisant la méthode Sanger. Ceci a ouvert la voie à un nouveau champ de recherches dans lequel
la dimension évolutive prenait de plus en plus de place par rapport à la dimension fonctionnelle. Les
nouvelles méthodes de séquençage ont considérablement accéléré le phénomène. Plusieurs dizaines
d'espèces de levures ont été séquencées, (Souciet et al., 2000, Wood et al., 2002, Cliften et al., 2003,
Kellis et al., 2003, Jones et al., 2004, Dujon et al., 2004, Dietrich et al, 2004, Kellis et al., 2004, Loftus
1 Dans un séquençage aléatoire, la couverture est donnée par le nombre de nucléotides totaux séquencés rapporté à la taille du génome. Si L
est la longeur moyenne (en nucléotides) de chaque lecture, N le nombre total de lectures effectuées et G la taille du génome (en nucléotides), la
couverture C s'exprime par C= NL/G). On a l'habitude d'exprimer ce rapport par un nombre de X (ex. 3X: couverture typique d'un séquençage
exploratoire, 6X: couverture typique d'un brouillon assemblé de séquence (draft), 10 -12 X: couverture standard d'une séquence qui sera
soumise à finition). Tous ces chiffres correspondent aux séquençages génomiques réalisés selon la méthode de Sanger jusqu’en 2007 environ.
Avec l'arrivée des nouvelles technologies, des couvertures beaucoup plus élevées sont obtenues et le problème des finitions est abandonné
faute de pouvoir le traiter (voir chapitre).
8et al., 2005, Dujon, 2005b, 2006, Novo et al, 2009, Dujon, 2010), et autant de champignons divers
(Galagan et al., 2003, 2005, Machida et al., 2005, Nierman et al., 2005, Dean et al., 2005, Kaiper et al.,
2006, Martin et al., 2008, 2010, Ma et al., 2009). On a séquencé des microsporidies (la première était
Encephalitozoon cuniculi, Katinka et al., 2001), des parasites comme le Plasmodium falciparum
(Gardner et al, 2002), agent de la malaria et son cousin P. yoelii yoelii (Carlton et al., 2002) et d'autres
Apicomplexes comme Cryptosporidium hominis (Xu et al., 2004), les trypanosomes Trypanosma
brucei (Berriman et al., 2005) et T. cruzi (El-Sayed et al., 2005), la leishmanie Leshmania major (Ivens
et al., 2005), des amibes comme Entamoeba histolytica (Loftus et al., 2005) ou Dictyostelium
discoideum (Eichinger et al., 2005) etc ... A mesure que l’efficacité de séquençage augmentait, la
génomique évolutive a pu également s’adresser aux organismes pluricellulaires. Douze espèces de
Drosophiles ont été séquencées et comparées pour comprendre l'évolution de ce groupe d'insectes
(Drosophila 12 genomes consortium, 2007). Le point critique était l’existence de centres de
séquençage capables de générer et de traiter des grands volumes de données.
Le Génoscope
En France, le Génoscope d'Evry, qui n'est pourtant que d'une taille modeste vis-à-vis de ses
concurrents étrangers, a réalisé le séquençage complet du chromosome 14 humain (Heilig et al.,
2003), du poisson Tetraodon (Jaillon et al., 2004), de la Paramécie (Aury et al., 2006), de la vigne
(Jaillon et al., 2007), d'une algue brune Ectocarpus silicosus (Cock et al., 2010), de l'urochordé
Oikopleura (Denoeud et al. 2010), pour ne citer que les plus grands projets. Depuis une quinzaine
d’années, il a réalisé plusieurs centaines de projets génomes au service de la communauté scientifique
française et européenne, en plus du séquençage de régions génomiques d'intérêt particulier, de la
recherche de mutations, de banques d'ADN complémentaires etc ... Aujourd’hui, le Consortium France
Génomique coordonne les activité de génomique en France.
Les curiosités biologiques
Avec les génomes, la Biologie traditionnelle redevient d'actualité. Par exemple, on a séquencé
les nucléomorphes de symbiontes récents tels que Guillardia theta, une Cryptophyte considérée à tort
comme une algue rouge (Douglas et al., 2001) et Bigelowiella natans, un Chlorarachniophyte considéré
à tort comme une algue verte (Gilson et al., 2006). Ces nucléomorphes représentent en réalité les
restes des noyaux d’algues rouge ou verte, respectivement, après leur absorption par d’autres
eucaryotes unicellulaires ayant ainsi acquis la photosynthèse de manière endosymbiotique (Curtis et
al., 2012). De la même façon, on a séquencé le génome d'une ascidie, Ciona intestinalis pour explorer
la base évolutive des Chordés (Dehal et al., 2002). On s'intéresse aussi aux annélides et aux
mollusques car ce sont des Lophotrochozoaires, une branche animale longtemps inexplorée au niveau
génomique et qui présente de nombreuses caractéristiques intéressantes au plan de la formation du
corps. Ou encore aux rotifères bdelloïdes, minuscules métazoaires asexués et résistants à la
dessiccation, dont le génome partiellement tétraploïde et hybride, montre une structure entièrement
nouvelle avec les deux allèles du parent diploïde portés par le même chromosome, inimaginable
d’après la génétique mendélienne classique (Flot et al. 2013). Loin d'être une activité réductionniste à
l'extrême comme certains l'imaginent, l'étude des génomes ouvre des voies nouvelles, d'une efficacité
inconnue auparavant, pour tous ceux qui connaissent l'Histoire naturelle et ses remarquables
observations. On s'intéresse aux symbioses, au parasitisme, et à toutes les interactions des
organismes dans la nature, dont la formation de nouveaux pathogènes.
Génomique populationnelle et métagénomique
9De plus, pour un nombre croissant d’organismes on séquence, pour les comparer, de nombreux
individus d’une même espèce. On parle de re-séquençage. C’est évidemment le cas pour l’homme,
mais aussi pour de nombreux microorganismes (voir par exemple Liti et al., 2009). Avec cette stratégie,
la génomique rejoint la génétique des populations, en l'enrichissant d'une quantité de données que
cette dernière ne pouvait pas obtenir par les méthodes traditionnelles. C'est là, l'un des défis majeurs
de l'enseignement de la Biologie moderne, tant ces disciplines sont restées trop longtemps séparées
(voir Lynch, 2007). De même, l'analyse des génomes nous affranchit de la nécessité d'isoler les
organismes étudiés, ce qui n'est pas toujours possible. Au contraire, on peut s'intéresser directement à
des populations naturelles, ou même des écosystèmes. On parle de métagénomique. Actuellement,
on découvre plus d'espèces nouvelles par le séquençage métagénomique que par les méthodes
traditionnelles. L'étendue de la biodiversité des espèces devient accessible aux nouvelles méthodes de
séquençage (Sogin et al., 2006). Les océans deviennent des champs d'exploration systématique. Un
projet piloté par des équipes françaises et le Génoscope (Tara Océans) a été lancé pour cataloguer
des virus, des bactéries et des eucaryotes unicellulaires des océans du monde entier (Karsenti et al.,
2011). Plusieurs centaines de prélèvements ont été effectués et les échantillons sont caractérisés par
le séquençage et l’analyse des morphologies cellulaires (Karsenti, 2012). Les échantillons océaniques
montrent de nombreux virus dont l’importance écologique et évolutive est probablement beaucoup plus
grande qu’on ne l’imaginait (Hingamp et al., 2013). A titre d’exemple, on se rappellera que les
Mammifères sont devenus placentaires à l’époque Crétacé par capture de gènes d’enveloppe
rétroviraux (Cornelis et al., 2014). Les sols aussi sont évidemment étudiés pour leur importance
agronomique ou forestière mais également pour suivre les effets de diverses pollutions (Monier, et al.,
2011). Au fur et à mesure que les résultats arrivent, on mesure l'ampleur de ce qui nous reste à
découvrir, même dans des systèmes limités comme les flores intestinales de l'homme ou des animaux
pour lesquels des programmes internationaux ont déjà livrés leurs premiers résultats (Qin et al., 2010).
On parle maintenant couramment de microbiome pour désigner les flores microbiennes dont les
compositions peuvent maintenant être intégralement décrites par la métagénomique sans nous limiter
aux micro-organismes cultivables. il y a actuellement plus de 11 000 métagénomes séquencés, qui ont
permis l’assemblage de près de 1 800 génomes d’organismes non cultivés (voir par exemple Bolotin et
al. 2014). C’est une véritable révolution par la puissance d’analyse déjà offerte à présent, qui devrait
encore être amplifiée par l’application de méthodes de séquençage incluant l’organisation
tridimensionnelle des génomes (Chromosome Conformation Capture et méthodes dérivées).
La phylogénomique
Enfin, c'est tout l'arbre du vivant qui est revu (et souvent corrigé) avec les données des
génomes. Il suffit pour s'en convaincre de regarder l'arbre actuel des eucaryotes (Baldauf et al., 2003,
Keeling et al., 2005, voir figure 3) et de le comparer avec les versions antérieures, même relativement
récentes. A la phylogénétique succède une phylogénomique dont les principes sont encore objet
d'actives recherches, vu la complexité du problème. La congruence des topologies des arbres devient
un problème très compliqué si l'on souhaite y intégrer toutes les données des génomes. Les arbres
obtenus dépendent du lot de gènes utilisé pour établir la phylogénie. Les raisons de ce phénomène
sont complexes et encore mal comprises. Les hybrides naturels et les transferts génétiques
horizontaux sont probablement beaucoup plus fréquents qu'on ne l'imagine comme le montrent les
microorganiemes.
Chez les bactéries, on constate que de nombreux segments de génomes varient entre isolats
d'une même espèce, reflets d'intenses échanges génomiques au sein des populations. La notion
même d'espèce s'estompe. On en vient à considérer un génome bactérien en deux parties, le "cœur"
10formé des gènes à transmission verticale (donc propres à la phylogénie) et les "ajouts" reflets d'une
intense dynamique horizontale. Les propriétés biologiques de l'organisme, ses capacités à s'adapter à
des niches écologiques ou, par exemple, à devenir pathogènes, sont la résultante finale des deux
parties (Danchin et al., 2007). Évidemment, certains organismes, dont l'homme, ont une reproduction
sexuée obligatoire, structurant les populations selon les lois de la génétique classique. Mais beaucoup
d'autres, surtout les microorganismes ou les champignons mais aussi les plantes ou même certains
animaux, ont des phases d'expansion clonale considérable dont on retrouve la signature dans les
génomes. Avec la perte fréquente de la sexualité dans de nombreuses lignées de microorganismes
eucaryotes, la notion d'espèce s'estompe encore plus.
Le problème de l’échantillonage taxonomique
A mesure que se précise l'arbre du vivant, on réalise à quel point nos connaissances actuelles
sur les génomes sont biaisées. Si l'on reporte les nombres de génomes connus sur les différentes
branches évolutives des eucaryotes, on s'aperçoit que l'essentiel des données correspond à deux
grandes divisions évolutives, celle des Opisthokontes qui rassemble tous les animaux et les
champignons et celle des Viridiplantae c’est-à-dire les plantes et les algues rouges et vertes. Si un
nombre raisonnable de données existent pour les Chromalveolata regroupant les Apicomplexes, les
Ciliés, les Algues brunes, les Oomycètes et quelques autres lignées, en revanche très peu, voire
pratiquement rien, est connu des génomes des deux autres grands groupes, Excavata et Rhizaria,
alors que les rares données disponibles suggèrent que beaucoup de surprises nous attendent. Les
modes actuels de financement de la recherche ne sont pas étrangers à ce phénomène. En privilégiant
la recherche finalisée, on n’étudie que ce que l’on est capable d’imaginer en se privant des surprises
importantes. Or la véritable recherche consiste à étudier ce que l’on ne connaît pas déjà !
11Viridiplantae+ Excavata+
2313$ 84$
8$
748$ Rhizaria+
5666$
Chromalveolata+ Unikonts+
Figure 3 : L'arbre phylogénétique des eucaryotes compte neuf lignées principales regroupées ici en cinq branches majeures (Keeling et
al, 2005). Le nombre de projets génomiques (rouge gras) répertoriés dans Genome On Line database (GOLD, octobre 2014) montre un fort
déséquilibre entre les cinq principales branches. La génomique a encore un long travail d’exploration à faire avant qu’une description
équilibrée du monde vivant ne devienne disponible.
Les nouvelles méthodes de séquençage
La période Sanger (1977-2007)
La méthode de Sanger était basée sur la synthèse in vitro de copies d'ADN complémentaire à
un brin matrice par les polymérases. La méthode de Maxam et Gilbert était basée sur la dégradation
chimique des molécules d'ADN. Les deux méthodes impliquaient le marquage terminal des molécules
et leur séparation selon leur taille par électrophorèse à haute résolution. Toutes les molécules d'une
même réaction de séquençage ayant une extrémité commune (origine) et l'autre dépendant de la
nature du nucléotide terminal, en les séparant par la taille, on lisait la séquence. Malgré leur apport
considérable à la Biologie, les méthodes initiales de séquençage ne permettaient pas une
augmentation d'échelle significative car elles nécessitaient trop d'interventions manuelles. Plusieurs
perfectionnements techniques, couplés aux progrès parallèles de l'informatique, allaient graduellement
changer le paysage jusqu’au milieu des années 2000. On peut citer la mise au point, puis l'utilisation
de nucléotides fluorescents qui, couplée à l'électrophorèse capillaire, allait permettre la construction
de toute une génération d'automates (séquenceurs) dont certains existent encore aujourd'hui dans
certains laboratoires où ils sont confinés à des tâches spécialisés. Avec les machines les plus
puissantes de cette génération technologique, on pouvait déterminer en parallèle 96 séquences
d'environ 750 nucléotides de long chacune, soit environ 70,000 nucléotides par "run" de deux à trois
heures. Ce sont ces méthodes de séquençage appliquant les principes fondamentaux de la méthode
Sanger qui, associées à des développements informatiques adaptés permettant d'assembler, finaliser
et annoter de très grands génomes, ont permis l'extraordinaire développement de la génomique
jusqu’à il y a quelques années.
12Les nouvelles méthodes de séquençage
Mais la situation a radicalement changée au milieu des années 2000 (voir par exemple, Seo et
al., 2005, Margulis et al., 2005, Shendure et al., 2005) avec l'arrivée de nouvelles méthodes de
séquençage souvent appelées NGS (pour Next Generation Sequencing). Contrairement aux
perfectionnements techniques précédents ces nouvelles méthodes appliquent des principes différents
de ceux des méthodes historiques. Elles ont été rendues possibles autant par les progrès de la
biologie moléculaire (nouvelles molécules, nouvelles réactions) que par ceux de l'ingénierie
(miniaturisation, traitement des images). Avec le NGS, la Biologie est entrée dans une nouvelle ère
pour plusieurs raisons. D’abord, les quantités de séquences produites sont beaucoup plus élevées que
celles obtenues par la méthode Sanger. Le pyroséquençage qui a ouvert cette période (maintenant
également lui-même presque abandonné) permettait en un seul "run" de lire un million de séquences
de longueur moyenne 500 nucléotides, soit un total de plus de 500 millions de nucléotides (à comparer
aux 70,000 nucléotides des méthodes précédentes). Avec les nouvelles méthodes utilisant la
synthèse en phase solide, un "run" peut produire plusieurs milliards de lectures de longueur de 100
nucléotides ou plus, soit un total de plusieurs centaines de milliards de nucléotides. C’est actuellement
cette dernière technologie qui est la plus utilisée dans le monde. Sa puissance est telle que, souvent,
plusieurs échantillons sont mélangés, après étiquetage moléculaire, pour être soumis à un
séquençage unique. Les séquences élémentaires sont ensuite aisément triées en utilisant les
étiquettes avant d’être traitées. C’est le volume des résultats de cette technique qui la rend
incontournable. Ses défauts sont la taille limitée de chaque séquence élémentaire, rendant les
assemblages problématiques en présence de séquences répétées ce qui est très souvent le cas, et le
besoin d’amplification des molécules par PCR avant le séquençage. D’autres techniques, basées sur
l'analyse de molécules uniques, permettent d’étudier les molécules d’ADN telles qu’elles existent dans
les cellules et non plus seulement leurs copies. Ces techniques sont encore en développement, bien
que déjà utilisées. Même si le volume de données produites n’atteint pas les performances dus
séquençage par synthèse en phase solide, elles offrent l’avantage d’allonger considérablement la
longueur de chaque lecture, point essentiel pour l'assemblage de novo de génomes inconnus, et de
réduire encore davantage le coût du séquençage. Il est vraisemblable que ces techniques seront
complémentaires les uns des autres.
La profondeur de lecture, élément critique
Avec ces nouvelles techniques, on entend souvent dire que le coût du séquençage a chuté en
quelques années de plus de 5 ordres de grandeur. Une performance rarement atteinte dans un
domaine économique ! C’est ce qui a permis au séquençage d’ADN de devenir une technologie
centrale pour de nombreuses applications (agronomie, environnement, cancer, génétique médicale,
recherche d’empreintes, criminologie etc …). On commence même à voir son application en routine
dans certains hôpitaux (en particulier pour l’analyse des tumeurs cancéreuses) et rien ne s’oppose à
étendre ces applications aux simples laboratoires d’analyse médicale. Il faudra ajuster les budgets de
santé en conséquence. Mais ce n’est pas cet aspect économique qui est le plus intéressant. Avec les
nouvelles techniques de séquençage, la multiplication des lectures est telle qu’elle permet enfin
d’atteindre des nombres comparables à ceux des molécules d’ARN dans une cellule ou au nombre de
molécules d’ADN d’un organisme pluricellulaire ou d’une population de microorganismes. L’étude
exhaustive se substitue à l’échantillonnage aléatoire. Ensuite, les méthodes NGS n'utilisent plus le
clonage de l'ADN dans des vecteurs d'E. coli qui fut la signature universelle du génie génétique depuis
plus de 35 ans et celle de la génomique pendant une quinzaine d'années. La séparation des molécules
d'ADN à séquencer et leur amplification se fait maintenant entièrement in vitro par PCR dans des
13micelles ou sur des supports solides. Avec les nouvelles technologies à molécules uniques, il n'y a
même plus d'amplification par PCR. Ce sont les molécules d'ADN présentes dans l'organisme étudié
qui sont directement séquencées. Avec l'énorme avantage de pouvoir identifier, en plus de la
séquence des 4 nucléotides fondamentaux, les modifications chimiques que ces molécules peuvent
porter et qui sont effacées par l'amplification par PCR.
Une révolution épistémologique
Evidemment, les bases de données et les logiciels d’analyse doivent s'adapter aux énormes
quantités de données produites par ces nouvelles méthodes. Il n'est plus envisageable de stocker les
données brutes de manière pérenne. Ces méthodes ont déplacé les limites des problèmes techniques
vers des problèmes d'informatique. Dans cette nouvelle Biologie qui émerge, la composition des
équipes de recherche et la formation de leurs membres, donc des étudiants, changent totalement.
L’effort d’analyse des données surpasse celui de la production des données. Mais les véritables
changements ne se limitent pas au volume des données à traiter. Le changement d'échelle induit un
changement de nature des questions étudiées. Les systèmes modèles traditionnels des laboratoires
(bactéries, levures, drosophile, souris, etc …) perdent de leur importance. Tous les organismes
existants deviennent étudiables. Ce sont leurs particularités biologiques qui font le degré d'intérêt de
leur étude. Les populations naturelles elles-mêmes deviennent accessibles à l'étude génomique, sans
se limiter aux espèces cultivables. L'évolution, les structures des populations, leur histoire, les forces
de sélection auxquelles elles ont été soumises deviennent lisibles dans les génomes. La génomique
de la biodiversité révolutionne notre connaissance des écosystèmes et des relations entre organismes
au sein de ces derniers. La métagénomique dépasse le catalogue existant d'espèces déjà identifiées
(très incomplet) pour nous ouvrir des mondes entièrement inconnus. Les ADN fossiles deviennent
analysables sans avoir besoin, d'abord, de les recopier en ADN moderne. En résumé, le changement
quantitatif a induit un changement qualitatif dans nos façons d’aborder la Biologie. En ce qui
concerne les applications médicales, quand chacun de nous aura son génome séquencé dès la
naissance (ou même avant), le problème restera celui de l’interprétation des données car les
déterminismes simples de type monogénique sont davantage l’exception que la règle et, de plus, le
polymorphisme génétique est beaucoup plus varié qu’on ne l’imaginait en étudiant seulement les SNP
dans les exons codants qui représentent moins de 2% de notre génome.
Retour sur les bases du système génétique
Avec le nouveau séquençage, la transcriptomique cesse d'être essentiellement quantitative
(mesure des quantités de transcrits par hybridation sur des arrays ou par séquençage d'étiquettes)
pour devenir analytique (les molécules d'ARN présentes dans une cellule sont séquencées
directement et quantitativement). Au lieu de se contenter de considérer les ARN comme de simples
intermédiaires de l'expression des gènes, ce sont les multiples formes de ceux-ci qui deviennent
analysables, y compris celles à courte durée de vie (Jacquier, 2009, Pelechano et al., 2013) qui
correspondent au fait que la transcription des génomes eucaryotes est générale et non limitée aux
gènes que l'on sait définir. Le séquençage massif d'ARN (par l'intermédiaire d'ADN complémentaire
soumis à séquençage massif) devient donc l'outil de choix pour annoter les génomes (Denoeud et al.,
2008). De nouveaux petits ARN non codants sont découverts. Et même les variations stochastiques
intercellulaires deviennent analysables grâce aux nouvelles méthodes de séquençage. (Newman et al.,
2006).
14Vous pouvez aussi lire

















































